VÝZKUM
2004
26.12.2004
PTC Therapeutics,
Inc. (PTC),
biofarmaceutická firma
zaměřená na výzkum, vývoj a komercializaci léků
oznámila zahájení I fáze studie PTC124 na zdravých dobrovolnících. PTC124 je
nový orálně podávaný lék zaměřený na nonsense mutace. S tímto lékem se
bude nejdříve zkoušet léčit cystická fibróza a Duchenneova svalová dystrofie,
ale do budoucna by mohl být použit pro řadu dalších genetických chorob. Předpokládá
se, že PTC124 by u DMD měl pomoci asi 5%÷15% pacientů.
·
Phase
I study of dystrophin plasmid-based gene therapy in duchenne/becker muscular
dystrophy
·
Systemic delivery of antisense
oligoribonucleotide restores dystrophin expression in body-wide skeletal
muscles
·
Should Foot Surgery Be Performed for
Children With Duchenne Muscular Dystrophy?
·
Coagulation system activated in
Duchenne muscular dystrophy patients with cardiac dysfunction
·
Muscular Dystrophy
Gene Therapy Trial Passes First Test by NIH Committee
·
Duchenne
Muscular Dystrophy: Stalled at the junction?
16.12.2004
·
2004
Annual Conference PPMD - Powerpoint Presentations
·
Duchenne Muscular
Dystrophy: Stalled at the junction? (pdf)
12.12.2004
Enzymový klíč k
úspěchu steroidů v léčbě DMD
|
|
|
|
Bernard
Jasmin |
|

Kortikosteroidní léky jako
Predisone nebo Deflazacort jsou často předepisovány pro zpomalení úbytku
svalové výkonnosti u Duchenneovy svalové dystrifie (DMD) a jiných
nervosvalových chorob. Oba pravděpodobně vedou ke zvýšené produkci bílkoviny
utrophin, která je podobná dystrofinu, což je bílkovina chybící u DMD. Zvýšená
produkce utrofinu může částečně kompenzovat chybějící dystrofin.
Nyní Lynn
Megeney (Ottawa Health Research Institute v Ontáriu), Bernard Jasmin
(Department of Cellular and Molecular Medicine, university of Ottawa) a
kolegové zjistili přesnou biochemickou signálovou cestu, která vede ke zvýšení
utrofinu.
|
|
|
|
Lynn Megeney |
|

Nový objev publikovaný 29. září
2004 v FASEB (Federation of American
Societies for Experimental Biology) ukazuje, že dystrofin deficitní buňky
potřebují enzym calcineurin pro regeneraci. Výzkum ukázal, že zvýšení
calcineurinu umožňuje dystrofin deficitním buňkám obejít biochemickou
signálovou cestou zvanou JNK1, která obvykle vede k jejich smrti, a že
zvýšení produkce utrofinu je jednou z hlavních výhod této signálové cesty.
“Naše pozorování nyní vyjasňují mechanismus, kterým steroidy dosahují
účinku“, říká Megeney. „Tato studie také potvrzuje, že patologie
dystrofin-deficitních svalů pochází částečně ze zvýšeného JNK1 a že
farmakologická korekce této signálové cesty upregulací calcineurinu je správný
přístup k léčbě DMD.“
Menegey dále uvádí, že vedlejší
účinky kortikosteroidů jako jsou zvýšení váhy, oční zákal, řídnutí kostí nebo
psychické problémy jsou závažnou překážkou pro jejich užívání. S novými
informacemi jakým mechanismem kortikosteroidy opravdu pomáhají dětem s DMD
je možné vyvinout lepší, přesněji cílené léky s redukovanými vedlejšími
účinky.
11.12.2004
·
Strand
bias in oligonucleotide-mediated dystrophin gene editing
·
Progress
in the treatment of Duchenne Muscular Dystrophy
7.12.2004
Nalezeny
kmenové buňky schopné přeměny ve svalové
Yvan Torrente z
Milánské University v Italy a kolegové v Itálii a Franci objevili způsob jak
identifikovat kmenové buňky cirkulující v krevním oběhu se schopností přeměnit
se ve svalové buňky. Giulio Cossu z Stem Cell Research Institute v San Raffaele
Hospital v Milan, který provádí výzkum v oblasti velmi blízké a je podporován MDA,
byl členem tohoto týmu.
Objev ukázal, že buňky
cirkulující v lidském krevním oběhu, které mají na povrchu protein nazývaný AC133,
se mohou přeměnit ve svalové buňky
pokud jsou v laboratoři vystaveny jisté látce nebo pokud jsou injikovány přímo
do tepen myší. Když vědci injikovali lidské kmenové buňky s AC133 do myší s
chorobou připomínající DMD přinejmenším některé se přeměnily ve svalové. (Myši
měly imunitní deficit umožňující jim přijmout lidské buňky.) Některé AC133
buňky se přeměnily v satelitní buňky, které se nachází okolo svalových vláken a
jsou schopny je opravit v případě potřeby. Ostatní buňky se přeměnily přímo ve
svalové.
“Protože tyto
buňky mohou být izolovány z krve, zpracovány in vitro a prostřednictvím
krevního oběhu v těle opět rozšířeny, představují možný nástroj vhodný pro
budoucí buňečnou terapii nejen DMD ale i některých jiných svalový dystrofií”
tvrdí badatelé ve své práci publikované v červencovém čísle Journal of Clinical
Investigation.
5.12.2004
·
TAMOXIFEN increases muscular
strength of the mdx dystrophic mice
·
HSCs for
muscular dystrophy: a paradigm shift, back
·
Duchenne
muscular dystrophy with associated growth hormone deficiency
·
Erythrocyte
from Duchenne muscular dystrophy is fragile
28.11.2004
·
Deleteriuos effects of
immobilization upon rat skeletal muscle: role of creatine supplementation
·
CAPON expression in skeletal muscle
is regulated by position, repair, NOS activity, and dystrophy
·
Mivacurium in Children
with Duchenne’s Muscular Dystrophy: A Comparison of Onset and Duration of
Neuromuscular Block with Children
without Neuromuscular Diseases
17.11.2004
·
Systemic delivery of
genes to striated muscles using adeno-associated viral vectors
·
Abstracts presented
on 14th Meeting of the European Neurological Society – Spain, June, 2004
o
Effect of
treatment with PDTC and IRFI 042 on strength and fatigue in MDX mice
o
Language
disorders in Duchenne's muscular dystrophy
14.11.2004
·
Development of an Innovative
Technique of Gene Therapy: Exon Skipping
Francouzská laboratoř Genethon (http://www.genethon.fr) založená a financovaná Association Francaise contre
les Myopathies (AFM, http://www.afm-france.org/) oznámila, že uspěla při léčbě svalů u mdx myší (myší
model DMD). Tým vedený Olivierem Danosem a Luisem Garciou ve spolupráci s
pařížským Cochin Institute toho dosáhl pomocí metody zvané “exon skipping”. Pro
přenos potřebné molekuly do svaly byl použit AAV (Adeno
Associated Virus) vektor. Tuto metodu bude možno
použít i pro léčbu některých jiných svalových dystrofií, cystické fibrózy nebo
třeba hemofilie. AFM teď rozjíždí rozsáhlý program na vyhledání dalších chorob
léčitelných touto metodou.
Laboratoře AFM připravují klinické pokusy pro DMD, sarcoglycanopathy,
calpainopathy a Wiskot-Aldrichův syndrom.
Do roku 2007 by měla být připravena první fáze klinických pokusů, u DMD
se zaměřením na exon 51. Podle Advances in
Duchenne muscular dystrophy „vystřižení“ exonu 51 by mělo vést k léčbě 17,5% delecí, což je největší
procento delecí léčitelné odstraněním jediného exonu. A zvládnutí odstranění
jen 6 exonů by mělo vést k léčbě 85% delecí u lidí (databáze: JC Kaplan,
Cochin Institute).
·
Researchers
make muscular dystrophy breakthrough
Bombasticky napsaný
článek nicméně s racionálním jádrem. „Genetic band-aid“ je jen jiný název
pro „molecular patches“ či „exon skipping“, čili jde o stejnou metodu jakou se
v Evropě zabývá tým Judith Van Deutekom. Brett Tizard slibuje, že další informace by se měly brzy
objevit na stránkách : http://www.anri.uwa.edu.au/. Bližší
informace Tizard slibuje prezentovat na konferenci “Muscular Dystrophy 2005”,
která proběhne 15. a 16. července 2005 v Perth v Austrálii. Více o této konferenci
: http://www.md2005.info/.
12.11.2004
·
Current concepts in
neuromuscular scoliosis
·
Evolving Therapeutic
Strategies for Duchenne Muscular Dystrophy: Targeting Downstream Events
·
Rescue
of Dystrophic Muscle Through U7 snRNA-Mediated Exon Skipping
·
Effects
of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx
mouse
·
Duchenne
Muscular Dystrophy research focuses on early mechanisms
·
Key clues
to muscle regeneration
·
Stem cell transplantation in DMD -
first report in China
V Číně
proběhl pokus s léčbou DMD pomocí kmenových. Asi před 3 měsíci byly
nitrožilně podány dvanáctiletému chlapci s DMD kmenové buňky získané
z pupeční šňůry nepříbuzného dárce. 76 dní po podání byla udělána svalová
biopsie, v některých svalech byl dystrofin slabě pozitivní a je jisté že
pochází z kmenových buněk. Chlapec vykazuje lehké zlepšení pohybových
funkcí. Další chlapec je připravován na transplantaci.
·
Abstracts presented on Annual
Meeting of The American Society of Human Genetics
o Therapeutic
antisense-induced exon skipping for Duchenne muscular dystrophy
·
Duchenne muscular dystrophy and dystrophin:
pathogenesis and opportunities for treatment
·
Helpful
Informations about Steroids in DMD
·
Muscle
ultrasound in children: Normal values and application to neuromuscular
disorders
11.11.2004
Tamoxifen
Ivana Švandová
Tamoxifen je cytostatikum, umělý nesteroidní
antiestrogen, který může s estrogeny cirkulujícími v krevní plasmě
kompetovat při vazbě na cytoplazmatický estrogenní receptor. Se slušnou
afinitou se na daný receptor naváže, pro „přirozený“ estrogen už tam pak není
místo – ale daný komplex Tamoxifen/receptor není funkční. Po přenesení do
buněčného jádra potom nedojde k rozmnožení proliferující nádorové buňky,
protože tento komplex zablokuje
spuštění přepisu některých genů tím, že se naváže například na jejich
promotorové sekvence (viz jak vzniká protein). Základem účinku Tamoxifenu je
tedy jeho vazba na estrogenový receptor, který už ale s tímhle ligandem
nemůže splnit svoji funkci, a místo aby např. stimuloval po vstupu do jádra
dělení buněk, tak mu naopak zabrání.
V případě toho typu rakoviny je sice tamoxifen
popisován jako antiestrogen, ale může se chovat taky jako agonista estrogenů
(napodobovat jejich účinek), nebo i obojace - částečně jako agonista nebo
antagonista, což závisí od studovaného živočišného druhu, cílového orgánu nebo
genetického potenciálu (na co se má účinkem zaměřit). Trochu kaše. :)
A jak to souvisí s TNF-b?
Tumor-nekrotizující faktory (TNF) jsou důležité
cytokiny – molekuly zapojené zejména do zánětlivých procesů. V tkáních (a
svalová není vyjímkou) se účastní reakcí spojených s chronickými stavy i
s akutním poškozením. Do poškození tkáně vcestují leukocyty a začnou
produkovat množství cytokinů, což posílí zánětlivou reakci, bez níž by následná
obnova tkáně nebyla možná (nejdřív je nutno zlikvidovat to poškozené). Hlavním
mediátorem zánětlivých reakcí bývá TNF-a, nazývaný taky kachektin.
Aktivuje chemotaxi leukocytů do místa zánětu, zvyšuje produkci adhezívních
molekul na povrchu dalších pomocných buněk, aby se mohly lépe „přilepit“ do
místa zánětu a pomáhat leukocytům, zvyšuje sekreci dalších prozánětlivých cytokinů
(interleukinu-1, interferonu gama), které pak posílí zánětlivou reakci. Tenhle
TNF má bratříčka TNF-b nazvaného lymfotoxin. Jeho
účinek není tak extrémní jako u TNF-a, ale váže se vlastně na tytéž
receptory. V případě chronické nebo nadměrné produkce TNF už není ovšem
činek zánětlivé reakce benefitem, ale škodí, začne být napadána i tkáň, která
si to zatím „nezasloužila“.
O estrogenech se ví, že potlačují produkci u TNF-a, a to tím, že potlačují jeho transkripci právě složitou soustavou reakcí
končící navázáním komplexů s estrogeny na promotor u TNF-a genu. Snižují tak možnost poškození tkáně přehnanou zánětlivou reakcí. O
tamoxifenu a některých dalších umělých anti/estrogenech se taky ví, že
potlačuje „dospívání“ určitého typu makrofágů (buněk zodpovědných za
nespecifické imunitní reakce) a brání jejich vyzrání na tzv. dendritické buňky,
které produkují protilátky. Je proto podáván jako podpůrná látka při některých
autoimunitních chorobách (hlavně kloubních nebo kožních - revmatické arthritidě,
lupence apod.). Obecně tedy tyto látky snižují sílu zánětlivých procesů.
Jak přesně a konkrétně interaguje tamoxifen se
systémem tumor-nekrotizujících faktorů nevím, ale pravděpodobně cestou snížení
jejich produkce zablokováním promotoru jejich genů. Zřejmě tedy výrazně snižuje
úroveň zánětlivých procesů ve svalu. Když pak zajde na zánět míň myocytů, které
by mu padly jen tak „bokem“ za oběť, bude i celý sval vykazovat lepší silové
vlastnosti.
13.10.2004
·
Tamoxifen increases muscular
strength of the mdx dystrophic mice
Tamoxifen
působí na TGF beta, což je skupina proteinů účastnící se degenerace a
regenerace svalů. V této studii byl tamoxifen podáván mdx myším a to v množství
5mg/Kg a 10 mg/Kg. Po pěti týdnech podávání byl zjištěn významný nárůst svalové výkonnosti a trval
po celou dobu podávání léku. Nyní se připravují pokusy na psech.
Tamoxifen
je lék běžně používaný na léčbu rakoviny prsou a bývá ženám podáván dlouhodobě
bez vážnějších vedlejších účinků. U mladších pacientů bývá podáván jen vzácně,
žádné vedlejší účinky nebyly pozorovány. Někdy bývá používán i na zánětlivé
choroby.
Experiment uskutečnil tým Dr. Davida
Federa z brazilského Genetic Therapy Center (CTG) v Ribeirao Preto. Toto centrum patří
brazilské Association of Friends of DMD (AADM) a bylo vybudováno a vybaveno z
darů rodičů a nadace State of São Paulo Research Foundation (Fapesp).
Podle informací od Dr. Davida Federa má jeho tým vytypováno asi 5 dalších
léků používaných na jiné choroby u kterých předpokládají pozitivní vliv na DMD a chystají se je v
nejbližší době začít testovat. Rychlost výzkumu je však silně limitována
omezenými finančními možnostmi. Dr. David Feder je otcem
devatenáctiletého syna Leonarda trpícího DMD.
·
Visualization of Ectopic
Calcification in mdx Mouse Skeletal Muscle
· Muscle
Proteoglycan Levels in Duchenne Muscular Dystrophy Differ from Other Muscular
Dystrophies
·
Sex Differences in Muscle Stem Cell
Transplantation Efficiency
7.10.2004
·
Dystrophy cure efforts rewarded
·
Third Round Table
Conference in Monaco on 19 June 2004
3.10.2004
·
Parent Project UK, Muscular Dystrophy
Campaign, 2004 Annual Conference
Ve
dnech 25 a 26 září proběhla konference PPUK.
Na této konferenci bylo oznámeno zahájení klinických pokusů s AO (Antisense Oligonucleotide) v
Británii a
Holandsku ještě do konce roku 2004. V první fázi půjde o testování bezpečnosti
AO a bude trvat přibližně 2 roky. V druhé fázi, která by mohla začít v roce 2006,
se bude zjišťovat účinnost AO. Bliží informace přineseme v nejbližších dnech.
·
Bone
Marrow Cells Regenerate Heart in Brazil Test
·
IGF1 Helps Leg Muscles in
DMD-Affected Mice
24.9.2004
·
Regenerative
capacity of the dystrophic (mdx) diaphragm after induced injury
·
Antisense
Therapeutics: A Promise Waiting to Be Fulfilled
·
Localization and early time course of
TGF-beta 1 mRNA expression in dystrophic muscle
·
'Mighty
mouse' helping find ways to prevent osteoporosis
16.9.2004
·
Heregulin
ameliorates the dystrophic phenotype in mdx mice, ( pdf )
·
'Mighty
mouse' helping find ways to prevent osteoporosis
15.9.2004
14.9.2004
PTC Therapeutics Inc.
biofarmaceutická společnost zaměřená na hledání, vývoj a komercializaci léků na
principu regulace produkce bílkovin malými molekulami oznámila dnes, že
obdržela grant 1.000.000$ od PPMD ( Parent Project Muscular Dystrophy ), aby
objevila nové látky pro léčbu DMD. PPMD a PTC společně vytypovali geny, jejichž
ovlivnění by mohlo mít terapeutický význam pro DMD. PTC použije svou
technologii GEMS ( Gene Expression Modulation by Small-molecules ) pro identifikaci malých molekul vhodných pro vývoj
nových léků pro léčbu DMD.
12.9.2004
·
Graffinity
Pharmaceuticals AG and MyoContract AG Merge to Form Santhera Pharmaceuticals Ag,
( pdf )
Výrobce léků, firma GRAFFINITY
a MYOCONTRACT AG se sloučily ve společnost SANTHERA PHARMACEUTICALS AG
Soukromá společnost GRAFFINITY Pharmaceuticlas AG a
MyoContract dnes oznámily své sloučení ve společnost SANTHERA PHARMACEUTICALS
AG. Tato nová společnost se zaměří na výzkum,
vývoj a komercializaci léku pro nervosvalová a metabolická onemocnění.
Sloučením biofarmaceutické společnosti zaměřené na nervosvalová
a metabolická onemocnění HEIDELBERG, Německo,
společnosti BASEL a LIESTAL, která soukromě vyvíjela léky AG a
MyoContractAG Švýcarsko vznikla nová
forma spolupráce SantheralPhramaceuticals AG. Finanční detaily nebyly
zveřejněny. Sídlo této nové společnosti bude ve Švýcarsku ( Liestal) výzkum a
vývoj bude probíhat také v Heidelbergu.
Postup Santery při vývoji léků zahrnuje i moderní
klinický program pro nervosvalové nemoci, který začne počátkem roku 2005 a dále
tři předklinické programy pro cukrovku, kachexii/anorexii a Duchennovu svalovou
dystrofii. Díky technologii, kterou vlastní Santhera bude možné uspíšit
programy spojené s vývojem nového léku. Díky vlastnictví této technologie je
Santhera vůdčí biotechnologickou společností v Evropě.
V důsledku zformování této jedinečné nové
společnosti směřují objevy předchozích společností ke komercializaci. Kombinace know-how Graffinity a MyoContractu vytváří
impozantní farmaceutickou společnost s
příslibem produkce nových léků pro nervosvalové nemoci s potenciálem v oblasti
metabolických poruch.
Sloučení vytvořilo jedinečnou příležitost vytvořit
silnější společnost s mnohem většími finančními zdroji, což umožní také s
produkty obchodovat.
Santhera Phramceuticals dnes stojí na prahu nové
éry. Vyvíjený lék se skládá z jednoho produktu ve fázi II/III klinického
ověřování pro použití u nervosvalových nemocí, jakož i pokročilý předklinický
vývoj zahrnující calpain inhibitory pro Duchenne svalovou dystrofii, inhibitory
pro cukrovku.
·
Mirus
Bio Enters into Research Agreement with Genzyme
·
Round table conference in Monaco on
19 June 2004: Transfer of the dystrophin gene
29.8.2004
·
Functional and Structural Improvements in
mdx Skeletal Muscle Following Chronic Daily Administration of Pyrrolidine
Dithiocarbomate (PDTC)
·
Effects of exercise and steroid on
skeletal muscle apoptosis in the mdx mouse
·
Administration of Insulin-Like Growth Factor-I
Improves Fatique Resistance of Skeletal Muscles from Dystrophic mdx Mice
·
Immune
Parameters
in Three Duchenne Muscular Dystrophy Patients Following Intramuscular
Transplantation of Normal Myogenic Cells Under FK506 Immunosuppression
24.8.2004
·
Experts Develop
Guidelines on Respiratory Care in DMD
·
Marathoning Mice
Could Have Olympian Effects on Obesity
·
Research
group unveils genetically-engineered mice
21.8.2004
·
Chimeric
RNA/Ethylene-Bridged Nucleic Acids Promote Dystrophin Expression in Myocytes of
Duchenne Muscular Dystrophy by Inducing Skipping of the Nonsense
Mutation-Encoding Exon (pdf)
·
Intraarterial
Delivery of Naked Plasmid DNA Expressing Full-Length Mouse Dystrophin in the
mdx Mouse Model of Duchenne Muscular Dystrophy (pdf)
·
Blood
loss in pediatric spine surgery
·
More information about
blood loss in DMD spine surgery
·
Newly
discovered protein may be key to muscular dystrophy
·
Respiratory
Care of the Patient with Duchenne Muscular Dystrophy - ATS Consensus Statement
·
2004
PPMD Annual Conference Powerpoint Presentations
·
A Facile Nonviral Method for
Delivering Genes and siRNAs to Skeletal Muscle of Mammalian Limbs
·
Targeted Exon Skipping in Transgenic hDMD
Mice: A Model for Direct Preclinical Screening of Human-Specific Antisense
Oligonucleotides
1.8.2004
·
Prednisolone
therapy in Duchenne muscular dystrophy prolongs ambulation and prevents
scoliosis
·
Deflazacort
in Duchenne muscular dystrophy: a comparison of two different protocols
·
Katie Bushby, UK, presentations :
o
General Principles of Management in DMD
o
Steroids — Speaking the Same Language
·
Respiratory
Care of the Patient With Neuromuscular Weakness: The (not so) New Paradigm
·
Prednisolone decreases cellular adhesion molecules required
for inflammatory cell infiltration in dystrophin-deficient skeletal muscle
31.7.2004
Vědcům podporovaným
MDA se podařilo doručit rozsáhlý gen do svalu
Vědci z
Univerzity ve Washington - Seattle doručili gen pro výrobu dystrofinu do
svalů DMD myší pomocí nitrožilní
injekce.. Přenos tak rozsáhlého genu dosud nebyl možný.
Šéf výzkumu Jeffrey Chamberlain (grant
MDA na Katedře neurologie, biochemie a medicíny) použil několik inovačních technik k doručení genu pro dystrofin,
který u DMD chybí a u BMD je nedostatečný. Valerie
Cwik, vedoucí lékařského výzkumu uvedla: “Hlavní cílem genové terapie pro
svalová onemocnění je zvýšení síly a funkce ve svalech. Současné výsledky
tohoto výzkumu dosažené na myších představují opět další krok k účinné léčbě
lidí". "Limitem v genové terapii do dnes byla skutečnost, že
nikdo dosud nenašel metodu, která by byla účinná pro všechny svaly dospělého
živočicha, včetně těch poškozených. Náš nový přístup je první, který umožňuje
doručení dystrofinového genu ke všem zasaženým svalům díky bezpečné a
jednoduché metodě."
Výzkum se liší od ostatních přístupů v
několika směrech :
1.
Vědci "zabalili" gen do nového typu
virového vektoru AAV6. Z viru jsou odstraněny všechny jeho vlastní geny a tak
je nemožné, aby se duplikoval v těle, zároveň se zdá být velice účinný a
bezpečný pro doručení genů do svalových buněk.
2. Vědci zvýšili schopnost nosiče s dystrofinovým genem tím, že myši podali
cévní endoteliální růstový faktor (VEGF). VEGF zvyšuje propustnost cév tak, že
nosič může pronikat do okolních svalů po té, co je vstříknut do krevního
řečiště. Použití této směsi by mohlo
být bezpečně použito i u lidí.
3. Vědcům se podařilo vyřešit problém nechtěné imunitní odezvy organismu.
Vědci zkoušeli dva různé molekulární " navaděče", které předají buňkám informace, aby začaly vyrábět
bílkovinu dystrofin.
Aktivita dystrofinového genu - výroba
dystrofinu byla patrná více než 8 týdnů. Hodnota kreatin kinázy ( enzymu, který ukazuje na
poškození svalových tkání) byla poloviční.
Nyní máme zásadní důkaz, že je možné do svalů
doručit nové, rozsáhlé geny.
Bezprostřední cíl je zjistit, zda to bude fungovat u lidí. Nyní plánujeme použití nového virového
vektoru na pacientech.
27.7.2004
·
Gene
Therapy Reverses Muscular Dystrophy in Animal Model
·
Systemic
delivery of genes to striated muscles using adeno-associated viral vectors
·
Annexin
expression in inflammatory myopathies
·
Ezrin-dependent
regulation of the actin cytoskeleton by {beta}-dystroglycan
·
Altered
activity of signaling pathways in diaphragm and tibialis anterior muscle of
dystrophic mice
·
Duchenne
or Becker muscular dystrophy in girls
·
Prednisone
reduces muscle degeneration in dystrophin-deficient Caenorhabditis elegans
·
Altered
blood-brain barrier development in dystrophic MDX mice
24.7.2004
·
Anesthetic Implications of
Neuromuscular Disease
·
Factors Influencing the Efficacy, Longevity, and Safety of
Electroporation-Assisted Plasmid-Based Gene Transfer into Mouse Muscles
·
Utrophin,
a way to cure Duchenne muscle dystrophy
·
Okadaic
acid augments utrophin in myogenic cells
18.7.2004
Farmaceutická společnost PTC Therapeutics startuje
I. fáze klinických pokusů s lékem nazvaným PTC 124, který byl vyvinut na speciální mutace v
dystrofinovém genu u chlapců s DMD. Pro bezpečnost bude testován na zdravých
dobrovolnících.
Pokud budou výsledky uspokojivé, plánuje
farmaceutická společnost v South Plainfield, N.J. druhou fázi na pacientech s
DMD v prvním pololetí roku 2005. (PTC
bude také testován na pacientech s cystickou fibrózou). " Jsme rádi, že se
tento projekt pohnul kupředu", řekla
Sharon Hersterlee, ředitelka výzkumu MDA. PTC 124 dokáže ovlivnit čtení
genetické informace, návodu pro tvorbu dystrofinu. To umožní přeskočit mutaci,
která by jinak zastavila toto čtení.
PTC 124 , lék, který je podáván ústně,
demonstroval schopnost obnovit tvorbu plně funkčního dystrofinu u myší s
DMD. Lék má podobné účinky jako
gentamicin, ovšem jinou strukturu a složení. Tento lék by mohl pomoci přibližně
15% pacientů s DMD, kteří mají tuto
specifickou mutaci v dystrofinovém genu. K určení této specifické mutace je
zapotřebí zvláštního testu, který je dostupný na Univerzitě v Utahu.
·
Decreased
total nitric oxide production in patients with duchenne muscular dystrophy
·
Tarantula
Venom Peptide Shows Promise as a Drug
14.7.2004
·
Therapeutic efforts in Duchenne muscular dystrophy;
the need for a common language between basic scientists and clinicians
· Investigation of Poor Academic Achievement in Children with Duchenne Muscular Dystrophy
· Depression in Parents of Children With Duchenne Muscular Dystrophy
10.7.2004
Biotechologická
firma Asklepios vyvíjí genovou terapii pro DMD/BMD
|
|
|
|
Xiao Xiao |
|

|
|
|
|
Virus nesoucí
DNA |
|
MDA přidělila částku 1.6 millionů USD Severokarolinské bitechnologické firmě Asklepios, aby vyvinula genovou terapii pro DMD/BMD. Badatelé Richard Jude Samulski, ředitel Gene Therapy Center na Univerzitě Severní Karolíny v Chapel Hill, a Xiao Xiao, biolog z Oddělení molekulární genetiky a biochemie pittsburské university již dříve pracující na grantech MDA, jsou členy výzkumného týmu ve firmě Asklepios.
Firma plánuje vyvinout a otestovat system doručování miniaturizovaného genu pro dystrofin do svalů chlapců s DMD za pomoci viru. Dystrofin je protein, který u pacientů s DMD svaly nedokáží vyrobit kvůli poruše na genu pro dystrofin.
Po rozsáhlém
toxikologickém odzkoušení vyžadovaném U.S. Food and Drug Administration (FDA),
firma plánuje provést 1. fázi
klinických pokusů na malém množství chlapců s DMD s cílem otestovat bezpečnost
této látky na lidech.
Pokrok
strategie čisté DNA pro léčbu DMD
|
|
|
|
Jon Wolff |
|

Jon Wolff grantový pracovník MDA na Pediatrickém oddělění na University of Wisconsin Madison byl v týmu, který oznámil na setkání Americké společnosti pro genovou terapii 2. až 6. července 2004, že byl učiněn významný pokrok u metody intravaskulárního doručení čisté DNA do svalů.
Wolff úzce spolupracuje s biotechnologickou firmou Mirus, jejímž je hlavním vědeckým poradcem. Wolff a kolegové, místo aby použili k dopravě genu do svalu virů, injikovali do žil končetin čtyř mdx myší úplný lidský gen pro dystrofin, tzv. čistou DNA. Končetiny zaškrtili, aby udrželi zvýšenou koncentraci genu ve svalech. Badatelé zjistili, že od 3% do 15% svalových vláken produkovalo dystrofin zatímco oni očekávali množství pod 0.5%. Horní svalové skupiny končetin vykazovaly vyšší výskyt dystrofinu.
Badatelé tvrdí, že tato metoda doručení genu do svalů se zdá bezpečná, afektivní a nevyprovokovává nežádoucí imunitní reakce narozdíl od virové metody, kde občas k takovým reakcím dochází.
Minulý rok AFM ( Francouzská asociace proti svalovým dystrofiím ) spolupracující s biotechnologickou firmou Transgene oznámila, že čistá DNA je účinná když je injikována do svalu (nikoliv do krevního oběhu). Pokusy proběhly na 15 chlapcích s DMD nebo BMD, bylo jim injikováno malé množství čisté DNA, které vedlo k produkci dystrofinu v 1% až 10% svalových vláken (Quest, červenec, srpen 2003).
6.7.2004
· Loss of Myostatin Gene Builds Muscle in Humans
·
Sizing up
muscular dystrophy
· Functional improvement of dystrophic muscle by myostatin blockade
3.7.2004
Minulý měsíc fermaceutická firma
Wyeth Pharmaceuticals začala první fázi pokusů s myo-29, látkou která blokuje
produkci myostatinu. Myostatin je protein regulující růst svalů. Studie
sponzorovaná firmou Wyeth publikovaná v New England
Journal of Medicine předpokládá, že blokování myostatinu by mohlo být
užitečné pro léčbu některých svalových chorob.
Publikovaná práce vychází ze
studia čtyřletého chlapce s dvojnásobným objemem svalů a polovičním množstvím
tuku než mají ostatní děti v jeho věku. Chlapec má zřejmě unikátní genetickou
mutaci, která potlačuje produkci myostatinu. Z tohoto ale i z dřívějších výzkumů
Wyeth věří, že blokování myostatinu pomocí myo-29 by mohlo pomoci při léčbě
některých svalových chorob. Firma se soustředí na možnost, že by lék mohl
zmírnit, ačkoliv ne vyléčit, svalové dystrofie. Dále by lék mohl být užitečný u
jiných chorob postihujících svaly nebo u svalové ochablosti související s
věkem.
Právě začala 1. fáze testování
myo-29 takže jeho uvedení na trh je ještě daleko. Myostatin blokující mutace
byly pozorovány jak v přírodě tak i nejméně u jednoho člověka a zatím nebyly
pozorovány negativními vlivy, proto by léčba potlačující tvorbu myostatinu
neměla mít vedlejší toxické účinky. To dává myo-29 šanci rychle přejít do fáze
2.
( Ve fázi 1 se testuje bezpečnost léku na skupině 20-80 lidí, ve fázi 2
účinnost a bezpečnost na 100-300 lidech, ve fázi 3 je lék podáván skupině 1000-3000 a je
vyhodnocová účinnost, monitorovány vedlejší účinky, lék je porovnáván s již
existujícími, pozn.překl. )
·
Myostatin Mutation
Associated with Gross Muscle Hypertrophy in a Child
·
Gene Mutation
Found in Muscle Man Toddler
·
Super-Strong
Toddler Studied For Muscle-Wasting Cure
·
Scientists
Study 'Baby Superman'
16.6.2004
·
Biotech
Firm to Develop Gene Therapy for Duchenne Muscular Dystrophy
MDA (Americká asociace svalových dystrofiků ) přidělila 1.6 milionu dolarů
severokarolinské firmě Asklepios pro
vyvinutí genové terapie pro DMD. Firma bude vyvíjet a testovat virovou metodu
dopravení miniaturizovaného genu do svalu chlapců s DMD. Dystrofin je svalová
bílkovina, která u této choroby chybí. Rozsáhlé testování bezpečnosti bude
vyžadováno před tím než bude možno testovat na lék chlapcích.
15.6.2004
Upregulace utrofinu
Ivana Švandová
Jedním
ze směrů, kterým
se ubírá vývoj léku proti DMD je upregulace čili zvýšení produkce bílkoviny
utrofinu. Utrofin je do značné míry strukturním
i funkčním homologem schopným dystrofin nahradit, a proto jsou všechny faktory
vedoucí ke zvýšení jeho exprese důležitým cílem pro event. terapii svalových
dystrofií. Pro lepší pochopení si nejdříve řekneme pár slov o tom jak vzniká
bílkovina a pak teprve o faktorech, které vedou k zvýšení utrofinu.
Jak vlastně vzniká bílkovina?
Velmi zjednodušeně pojato, každá bílkovina je
kódována v naší DNA, deoxyribonukleové kyselině, která se skládá z
„písmenek“ – tzv. bází. Pokud má vzniknout nová molekula proteinu zakódovaného
v DNA, musí se určitý úsek DNA tzv. přepsat procesem, kterému se anglicky
říká transcription. Vznikne messenger RNA (mRNA), „poslíčková“ RNA – molekula
RNA, ribonukleové kyseliny, která je tvořena taky bázemi, ale oproti DNA je to
už specificky úsek, který víceméně odpovídá proteinu, jenž má vzniknout. Tato
mRNA je dále „překládána“ v procesu translace: každé trojici bází odpovídá
kódovaní nějaké aminokyseliny, a aminokyseliny jsou základní kamínky, z nichž
jsou složeny proteiny. Během translace vznikne spojováním aminokyselin (podle
toho, jak jsou postupně na mRNA „čteny“ jednotlivé trojice aminokyselin)
bílkovina, je pak patřičně upravena a
přenesena tam, kde má plnit svoji funkci.
To, jaký vznikne kdy protein, je složitě řízeno – na
DNA jsou za sebou uloženy báze kódující rozdílné proteiny. Aby se to buňce
nepletlo, má každý protein svoji adresu, poznávací značku, tzv. promotor.
Promotor je určitý kousek DNA, který leží před bázemi kódujícími vlastní
protein. Pokud je tento promotor odblokován nebo přímo stimulován, dochází
k expresi daného proteinu, nebo alespoň jeho mRNA (v buňce lze detekovat
mRNA pro spoustu proteinů, jejichž zvýšenou expresi ale nezaznamenáme, tj.
dojde k přepisu, ale ne už k překladu).
Heregulin
Heregulin je růstový faktor, jehož podání zvyšuje
hladinu nových molekul utrofinu
v buňce. V tkáňových kulturách myotub (myších i lidských)
zvedlo podávání heregulinu hladiny mRNA pro utrofin 2.5krát. Obdobně došlo po
podání genu pro heregulin přímo do svalové tkáně ke zvýšení exprese genu pro
utrophin také v in vivo pokusech.
Heregulin je látka (malý protein), která nepřímo
reaguje s promotory různých genů. Prozatím je přesvědčivě doložen
vztah mezi heregulinem a utrofinem a
heregulinem a některými podjednotkami acetylcholinového receptoru (AChR), což
je nejdůležitější typ receptoru na nervosvalovém spojení. Hladiny utrofinu i
některých podjednotek AChR se zvýšily nejen v pokusech na tkáňových
kulturách, ale také in vivo. Po navázání heregulinu na příslušný
receptor dojde k aktivaci tzv. MAP-kinas, bílkovin navazujících fosfátovou
skupinu na další proteiny v buňce. Tím tyto proteiny aktivují a
v konečném důsledku dojde k aktivaci promotoru utrofinového genu. Gen
je přepsán, přeložen a vzniká nová molekula utrofinu.
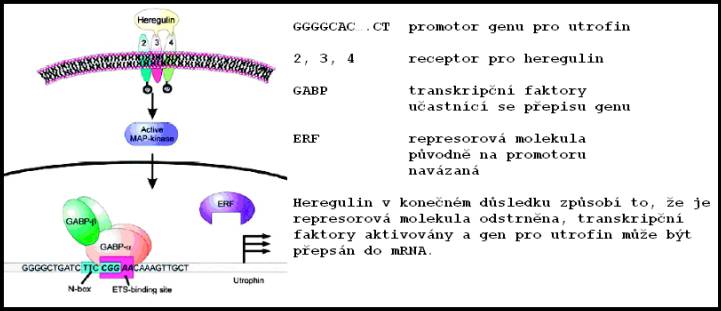
Zdroje: Molecular Biology of the Cell – odtud výřez
z obrázku (Vol. 10, 2075-2086, 1999), Nucleic Acid Research (Vol. 29, No. 23,
4843-4850, 2001) a PNAS (Vol. 96, 3223-3227, 1999).
L-arginin
L-arginin
je substrát enzymů ze třídy NO synthas. NOS jsou proteiny, které metabolizují
arginin: mění jej na jinou aminokyselinu, L-citrulin, za současného vzniku
molekuly oxidu dusnatého (NO, obr. 2). Ve svalových vláknech jsou NOS spojeny
s proteiny dystrofinového komplexu (DAPC). U dystrofických pacientů
jsou v nejhojnější míře
degradována aprávě tzv. 2b vlákna, která obsahují NOS.
Oxid
dusnatý má mnohé biologické účinky – je to malá a velice reaktivní molekula
(radikál), ochotně reagující se širokou škálou látek a proteinů v buňce.
Ve
svalové tkáni NO zvyšuje sílu kontrakce vlákna. NO je silné vasodilatans, jeho
působením se zvyšuje průsvit cév zásobujících sval – sval má tak větší přísun
živin (glukosy) a kyslíku a naopak produkty jeho metabolismu nebo degradace
jsou rychleji odváděny; obecně se dá říci, že napomáhá dolaďovaní vztahu mezi
metabolickými potřebami svalového vlákna a okolím. NO také přímo ovlivňuje
metabolismus glukosy a železa. Velkou hrozbou patologických procesů ve všech
typech tkání je vznik tzv. kyslíkových radikálů – NO je může „vychytávat“, reagovat s nimi a
snižovat tak jejich obsah v buňce, tedy snižovat riziko tzv.
lipoperoxidačního poškození membrány a poškození DNA (obr. 3). NO dále pomáhá
udržení klidového membránového potenciálu na „normálních“ hodnotách, což je pro
svalové vlákno signálem, že jeho komunikace s okolím je v pořádku, a
pravděpodobně velmi složitou signální kaskádou napomáhá zpětně kontrolovat
funkční stav nervosvalového spojení.
Celkově
se dá říci, že podáváním L-argininu jako substrátu NO synthas se výše uvedené
účinky NO posílí. Navíc se vyskytuje stále více dokladů, že metabolismus NO
(tj. i jeho zvýšená syntéza po přidání L-argininu) ovlivňuje i syntézu
strukturních proteinů nervosvalového spojení, hlavně utrofinu.
Zvýšená
hladina NO v organismu má i svá rizika: vyjma nepříjemných tzv.
nitrátových bolestí hlavy (způsobených hlavně rozšířením cévního řečiště) NO
prokazatelně zvyšuje riziko krvácení (inhibuje shlukování krevních destiček)
aj.
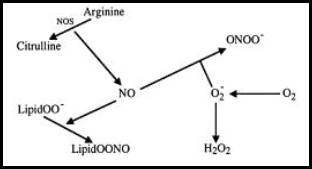
Obr.2 : Reakce NO
s dalšími aktivními kyslíkovými radikály.
Z Neuromuscular Disorders 11
(2001) 517-524.
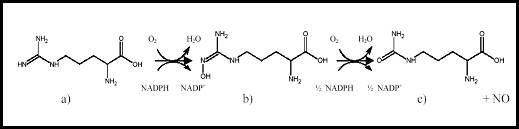
Obr.3 : Konverse L-argininu
na L-citrulin plus oxid dusnatý (NO).
a) L-arginin; b) NG-hydroxy-L-arginin;
c) L-citrulin.
ADAM 12
ADAM
12 (nazývaný také meltrin) je jeden ze zástupců proteinů tzv. ADAM (a disintegrin
and metalloprotease) rodiny. Jde o proteasu (bílkovinu štěpící
jiné bílkoviny), která podporuje uchycování buněk v tkáních, dále
diferenciaci a splývání myoblastů (nezralých buněk svalových
vláken), nebo také např. splývání vajíčka se spermií během oplodnění.
V lidském organismu se ADAM 12 vyskytuje ve dvou variantách: jako dlouhý
protein zakotvený v buněčné membráně (ADAM 12-L) a jako krátká,
secernovaná (buňkou do okolí vylučovaná) forma (ADAM 12-S). Je detekovatelný
zejména v placentě, u dospělých jedinců v tkáni hladké i příčně
pruhované svaloviny.
Svalové
vlákno kosterní svaloviny je útvar vznikající splýváním mnoha nezralých
svalových buněk – myoblastů. Tyto myoblasty
přilnou jeden k druhému nejprve okraji své membrány a postupně
splývají, až vznikne mnohajaderný útvar, svalové vlákno. Během vzniku svalového
vlákna (myogenese) byla v jeho membráně pozorována právě zvýšená exprese
ADAMa 12. Ve zdravé svalové tkáni se vyskytují tzv. satelitní buňky. To jsou
buňky, které se neúčastní hlavní činnosti svalu, kontrakce, ale slouží zejména
jako „zásobárna“, vznikají z nich nové myoblasty, když je sval poškozen a
je potřeba nahradit část tkáně. Zobecněně, ze satelitní buňky se stane
myoblast, a tyto myoblasty fúzují, až vytvoří nové svalové vlákno. Pokud by se
tyto buňky podařilo cíleně „nastartovat“ a nové myoblasty pak donutit
k fúzi, mohla by se otevřít další potenciální cesta k reparaci
svalové tkáně dystrofických pacientů.
Skupina
dr. Wewerové z Dánska použila upravené buňky nádorového onemocnění svalové
tkáně (myosarkomové buňky), do kterých vnesla gen pro krátkou formu ADAMa 12.
Tím byly buňky nuceny produkovat velké množství tohoto proteinu, a vědci
sledovali, zda buňky v tkáňové kultuře nezačnou produkovat myoblasty,
které by postupně splývaly za vniku svalového vlákna. Po nějaké době získali
směs buněk, v níž identifikovali myoblasty v různých stadiích
myogenese, a dokonce i fúzující myotuby s náznakem příčného pruhování,
typického pro vlákna kosterní svaloviny a pro svalovinu srdeční. Tyto buňky ale
nebyly uspořádány do žádného organizovanějšího útvaru. Nové myoblasty vznikaly
pravděpodobně z buněk satelitních, popř. z blíže nedifinovaných buněk
přítomné pojivové tkáně. V případě tohoto specifického typu myosarkomu se
tedy podařilo pomocí ADAMa 12-S navodit myogenesi.
Zdroj:
Novel, Secreted Form of Human
ADAM 12 (Meltrin a) Provokes Myogenesis in Vivo
Brent J. Gilpin, Frosty Loechel,
Marie-Genevie`ve Mattei, Eva Engvall, Reidar Albrechtsen, and Ulla M. Wewer
THE JOURNAL OF BIOLOGICAL CHEMISTRY
Vol. 273, 1, 157–166, 1998
Můj osobní názor je takový, že do
doby, kdy nebudeme víc rozumět procesům vedoucím ke splývání buněk a
k tomu, jak je donutit se dělit a spojovat cíleně, žádný klinický pokus
s ADAMem 12 nemůžeme čekat, protože s sebou přináší zejména příliš
veliké riziko neoplasií, nekontrolovatelného buněčného dělení v různých
typech tkání:
Práce dr. Wewerové pokročila
dále, jako první se ji na transgenních myších podařilo také dosáhnout toho, aby
produkovaly zvýšené množství ADAMa 12. Samičky těchto myší trpěly těžkou
nadváhou, ve svalové tkáni jim vznikalo velké množství tukových buněk
(adipocytů), tuk se ukládal i v dutině břišní. Samečci byli také
obtloustlí, i když celková míra těchto projevů nebyla taková jako u samic. Ke
vznikům nové tukové tkáně bylo zapotřebí dlouhé varianty ADAMa 12. Krátká
varianta (ADAM 12-S), která nemá proteasovou aktivitu, přispívala ke vzniku
adipocytů jen omezeně.
Zdroj:
DAM 12 Protease Induces Adipogenesis in Transgenic Mice
Nobuko
Kawaguchi, Xiufeng Xu, Rie Tajima, Pauliina Kronqvist, Christina Sundberg,
Frosty Loechel, Reidar Albrechtsen and Ulla M. Wewer
American Journal of Pathology. 2002;160:1895-1903
15.6.2004
·
Researchers
Report Major Advance In Gene Therapy Technique
·
Regenerative
capacity of the dystrophic (mdx) diaphragm after induced injury
·
MDA To Plan North
American Clinical Trials Network For Muscular Dystrophy
·
Recent advances in paediatric
muscular dystrophies
·
Progression
of scoliosis after spinal fusion in Duchenne's muscular dystrophy
·
Sacral
Nerve Stimulation for Treatment of Fecal Incontinence in a Patient With
Muscular Dystrophy
·
Cardiomyopathy in Muscular
Dystrophy Workshop 28–30 September 2003, Tucson, Arizona
10.6.2004
Kreatin pomáhá
u DMD
U dětí s DMD, které užívaly
kreatin jako dietní doplněk v dávce 0.1 gramu na 1kg váhy po dobu 4 měsíců se prokazatelně
zvýšila síla stisku vyhraněné ruky i objem svalové hmoty. Mark Tarnopolsky
(grant AMD), profesor pediatrie na Univerzitě v Hamiltonu, Ontario, Canada,
vedl studii s 30 chlapci ,jejichž průměrný věk byl 10 let. 15 chlapcům byl podáván kreatin po dobu 4 měsíců,
pak byla pauza 6 týdnů bez kreatinu a
pak dostávali placebo . Druhá polovina chlapců dostávala placebo, které později
nahradil kreatin. Plicní funkce, denní aktivita a schopnost plnit své úkoly - chůze do schodů, vystřihování,
neprokázaly žádné zlepšení ale
biochemický ukazatel degenerace se snížil. „Studie trvala pouze 4
měsíce. Bylo by třeba uskutečnit ještě dlouhodobější pokusy, abychom zjistili,
zda kreatin má účinek i na funkčnost svalů“, uvedl Tarnopolsky.
Ostatní studie s kreatinem u
různých neurologických nemocí přinesly rozporuplné výsledky. Belgická
studie (2003) s 15 chlapci DMD a BMD porkázala, že skupina, která užívala
kreatin měla méně kontraktur, větší sílu a nižší sklon k únavě, zvýšila se také
četnost chůze. Výsledky pokusů Tarnopolského i ostatních vědců ukazují na
příznivé účinky kreatinu. Tuto myšlenku potvrzují i studie na zvířatech.
28.5.2004
Vyjádření MUDr. Vondráčka ke klinickým pokusům s Cyclosporin-A a Prednisone
18.5.2004 jsme uveřejnili článek „Informace o klinickém pokusu s Cyclosporin A a Prednisone pro léčbu DMD
od Prof.Dr. Rudolfa Korinthenberga“. K tomuto článku jsme dostali
následující vyjádření MUDr. Petra Vondráčka z Ambulance Kliniky dětské
neurologie, Fakultní dětské nemocnice Brno :
Osobně jsem k jejich projektu spíše skeptický z následujících důvodů:
1. Oba medikamenty, které mají silné protizánětlivé a imunosupresivní účinky, mohou ve svalech potlačit zánětlivé změny, které tam bývají jako jeden z vedlejších důsledků DMD a tím snad krátkodobě zvýšit sílu některých svalů. Douhodobý efekt však nikdy zatím nebyl prokázán a potenciální vedlejší efekty jsou vyznamné ( potlačení přirozené imunity, poruchy tvorby kosti, zásah do hormonálních regulací ).
2. Jejich
studie se opírá o 2 uvedené studie z let 1993 a 1997, provedené v California
Pacific Medical Center v San Franciscu. Od té doby ( 7 let ) však ani oni ani
nikdo jiný o tom nic seriózního nepublikoval, ani v CPMC v tomto výzkumu dále
nepokračovali, což je jistě divné, kdyby to bylo perspektivní. Zdá se proto, že
se skutečně nejedná o aktuální perspektivní trend.
S pozdravem Dr.Vondráček
27.5.2004
· Therapeutic gene transfer to dystrophic diaphragm by an adenoviral vector deleted of all viral genes
· Cardiopulmonary failure in Duchenne muscular dystrophy--pathophysiology and management
· Decreased total nitric oxide production in patients with duchenne muscular dystrophy
·
23.5.2004
·
Creatine
monohydrate enhances strength and body composition in Duchenne muscular
dystrophy
·
Effect
of creatine supplementation on skeletal muscle of mdx mice
18.5.2004
Naše studie
začala v lednu 2004. Bude se na ni podílet 10 středisek v Německu, Švýcarsku a
pravděpodobně i Rakousku. Tato střediska budou muset najít 150 chlapců starších
pěti let. Během prvních tří měsíců léčby budeme porovnávat terapii
Cyclosporinem A (CsA) s placebem. Chceme vědět jestli dojde k nárůstu síly jak
se dá předpokládat z 2 předchozích studií. Během následujících dvanácti měsíců
budou obě skupiny ( CsA i placebo ) navíc léčeny pomocí Prednisone
v přerušovaném režimu. Tímto chceme zjistit jestli účinnost léčby pomocí
Prednisone může být zvýšena pomocí CsA. Dále, samozřejmě, chceme monitorovat
vedlejší účinky.
11.5.2004
·
Small-molecule
gene therapy nearing trials
·
Regeneration of
Injured Muscle from Adult Stem Cells
10.5.2004
·
Okadaic acid
augments utrophin in myogenic cells
·
Dystroglycan,
a scaffold for the ERK–MAP kinase cascade
8.5.2004
o
Micro-Utrophin as a Therapeutic
Protein in rAAV Mediated Gene Therapy for Duchenne Muscular Dystrophy
o
Gender Differences in
Transplantation Efficiency Using Muscle-Derived Stem Cells for Muscular
Dystrophy
o Therapeutic
Antisense-Induced Exon Skipping for Duchenne Muscular Dystrophy
o New
Canine Models of Duchenne Muscular Dystrophy: Identification and Molecular
Characterization
o A
Novel MiniDys-eGFP Fusion Gene for Developing Cell-Based Therapies of Duchenne
Muscular Dystrophy
o Expressing Full-Length
Dystrophin in 50% Cardiomyocytes Corrects Cardiomyopathy in the Mdx Mouse Model
for Duchenne Muscular Dystrophy
o Expression
of Normal Dystrophin Following Myoblast Transplantation to Duchenne Muscular
Dystrophy Patients
o rAAV-Mediated
Gene Therapy To Treat Limb Girdle Muscular Dystrophy Type 2D (LGMD-2D)
o Transgenic
Expression of Dp116 in Muscle Does Not Ameliorate Dystrophy in mdx4cv Mice
o Systemic
Gene Transfer to Striated Muscles Using rAAV6 Vectors
o Lentivirus
Mediated Dystrophin Expression in mdx
Muscles
o Nucleofection
and Phage phiC31 Integrase Mediate Stable Introduction of a Dystrophin Fusion
Gene into Muscle Derived Stem Cell and Human Myoblasts
o Successful
AAV Vector-Mediated Gene Transfer into Canine Skeletal Muscle Required
Suppression of Excess Immune Responses
o An
AAV Vector-Mediated Micro-Dystrophin Expression in Relatively Small Percentage
of Dystrophin-Deficient mdx Myofibers
Still Improved the mdx Phenotype
through Compensatory Hypertrophy
o Muscle-Derived
Stem Cells Display an Extended, but Not Unlimited, Expansion Capability:
Implication for Muscle Regeneration
o Delivery
of Igf-I and Dystrophin to Dystrophic mdx
Muscle
o Immunogenicity
of Dystrophins Delivered to Mice by Gutted Adenoviral Vectors
o Human
Myoblast Deimmortalization Using Tat-Mediated Cre Recombinase Delivery
o The
Fetal Approach: A Novel Therapy for the Treatment of Musculo-Skeletal Disease
o
In Utero Intramuscular Delivery of a High-Capacity Adenoviral
Vector Carrying a Full-Length Murine Dystrophin cDNA Provides Functional
Benefit and Reassembly of the Dystrophin-Glycoprotein Complex in Hind Limb
Muscles of mdx Mice
·
Glucocorticoid
corticosteroids for Duchenne muscular dystrophy
·
Selected articles
(22) from ATS - 2003,2002,2001 (American Thoracic Society International
Meeting)
·
Inducing
muscle hypertrophy as a therapeutic strategy for muscular dystrophies
·
Anti-TNFalpha
(Remicade) therapy protects dystrophic skeletal muscle from necrosis
17.4.2004
·
Researchers
pinpoint pathway involved in muscular regeneration
·
Inducing
muscle hypertrophy as a therapeutic strategy for muscular dystrophies
11.4.2004
Jak funguje Myodur
Ivana Švandová
Myodur
je nově vyvíjený lék na svalové dystrofie, který vyvíjí biotechnologická firma Xechem. Pro pochopení toho jak funguje je
potřeba si říct nejdříve pár slov o proteaze kalpain a vápníku.
Kalpain
Kalpainy
jsou rozsáhlá rodina proteas – enzymů štěpících jiné proteiny v buňce.
Štěpení (degradace) proteinů je proces normální, fyziologický, bez něj bychom
například nemohli strávit většinu toho, co sníme (trypsin ve šťávě slinivky
břišní je také proteasa).  Pokud
se ovšem proteas v buňce vyskytuje moc, jsou přehnaně aktivní nebo nejsou
včas odbourány (deaktivovány), vznikají problémy – například degradace svalové
tkáně. Různých forem kalpainů je asi 15; nejlépe prozkoumány jsou proteasy
μ-kalpain, m-kalpain a kalpain 3 (jiná jména kalpainu 3 jsou CAPN3, p94).
První dva se vyskytují ve všech buňkách savčího organismu, výskyt p94 je
specifický pro kosterní svalovinu. Všechny tyto formy jsou aktivovány
vápenatými ionty (na vápníku závislé, Ca2+ dependentní). Svalový
kalpain 3 rozkládá molekuly jako fodrin (spektrin II), desmin, filamen, nebulin, vimentin, gelsolin, and vinculin
C-protein, tropomyosin, troponin T, troponin I, titin, neničí ale přímo hlavní
molekuly kontraktilního aparátu: alfa-actin, alfa-actinin nebo tzv. těžké
řetězce myosinu.
Pokud
se ovšem proteas v buňce vyskytuje moc, jsou přehnaně aktivní nebo nejsou
včas odbourány (deaktivovány), vznikají problémy – například degradace svalové
tkáně. Různých forem kalpainů je asi 15; nejlépe prozkoumány jsou proteasy
μ-kalpain, m-kalpain a kalpain 3 (jiná jména kalpainu 3 jsou CAPN3, p94).
První dva se vyskytují ve všech buňkách savčího organismu, výskyt p94 je
specifický pro kosterní svalovinu. Všechny tyto formy jsou aktivovány
vápenatými ionty (na vápníku závislé, Ca2+ dependentní). Svalový
kalpain 3 rozkládá molekuly jako fodrin (spektrin II), desmin, filamen, nebulin, vimentin, gelsolin, and vinculin
C-protein, tropomyosin, troponin T, troponin I, titin, neničí ale přímo hlavní
molekuly kontraktilního aparátu: alfa-actin, alfa-actinin nebo tzv. těžké
řetězce myosinu.
Je to
užitečná molekula, bez její funkce by se ve svalovém vlákně hromadily proteiny
opotřebované zátěží, nicméně pokud není včas v patřičné míře deaktivována,
začne vlákno ničit.
Kalpain 3 (p94) má jednu
unikátní vlastnost – velice rychle rozkládá sám sebe (podléhá autolýze), čímž
se deaktivuje. V jeho genu bylo nalezeno víc než 100 různých typů mutací.
Většina z nich nezmění jeho strukturu natolik, aby nemohl dál štěpit své
substráty, ale pozmění jeho autolytickou funkci: zmutovaná molekula kalpainu 3
se sama nerozkládá s tou ohromnou rychlostí jako molekuly vytvářené na
podkladě nezmutovaného genu, časem se ve svalovém vlákně hromadí a díky tomu,
že jiné její aktivity jsou zachovány, jej ničí, nadměrně degraduje proteiny
potřebné k normální funkci vlákna.
To je
podstatou specifické formy svalové dystrofie, tzv. LGMD2A dystrofie. Zatímco
jiné formy dystrofií jsou dány hlavně primární poruchou tvorby strukturních
proteinů membrány svalového vlákna (hlavně dystrofinu, ale i dalších proteinů
stabilizujících membránu – sarkoglykanů, spektrinů aj.), tato forma dystrofie
je vlastně poruchou enzymovou – někdy se jí říká také kalpainopathie. Kalpainy
jsou obecně aktivovány vtokem vápenatých iontů do buňky. Tento tok je u všech forem
dystrofií zvýšený, protože membrána vlákna je strukturně nestabilní, takže
zvýšenou aktivitu kalpainů lze pozorovat u téměř všech dystrofiků. Toto je
ovšem efekt sekundární, nezapříčiněný snížením autolytické funkce kalpainu 3
při různých mutacích. Spousta pacientů s jinými formami dystrofií, než je
LGMD2A, má lehce defektní gen pro kalpain 3, takže sice trpí jinou dystrofií,
ale snížená autolýza kalpainu se k tomu ještě přidá…
Kromě
přímé zvýšené degradace proteinů také mutace v kalpainu 3 zvyšují náchylnost
k poškození membrány svalového vlákna pracovním stresem, brání řádné
myogenesi (normálnímu vzniku nových svalových vláken) a normální diferenciaci
vláken už zralých.
Vápník
Vápník
je nejdůležitější iont v buněčné signalizaci, jakákoliv živá buňka si
udržuje jeho koncentraci ve svém vnitřku asi o 4 řády nižší, než je
v jejím extracelulárním okolí. To dělá právě proto, aby si „všimla“ i malé
změny v koncentraci vápníku uvnitř. Vápenaté ionty se váží na spoustu
enzymů. Enzym je vlastně řetězec základních stavebních kamínků – aminokyselin,
které jsou buď nepolární, nebo různě nabité. Díky tomu, jak jdou za sebou, se
na základě náboje různě odpuzují nebo přitahují, a tak vzniká 3D struktura
enzymu, která je nesmírně důležitá pro jeho funkci. Velmi hrubě řečeno, pokud
není správná, nemůže se třeba dostat látka, co má být enzymem opracována, do
tzv. aktivního centra enzymu, a enzym nefunguje.
A jak
to souvisí s vápníkem? Vápník je nabitý, spolu se svým obalem tvořeným
vodou dost velký iont. Když se naváže na enzym, vždy změní jeho 3D uspořádání.
Většinu enzymů to aktivuje, některé naopak inhibuje, může také docházet
k otevírání iontových kanálů a spoustě dalších událostí. Enzymy nepracují
samostatně, ale v kaskádách, kdy činnost jednoho je závislá na předchozím.
Vtok vápníků tak může (nejčastěji) aktivovat ohromný sled reakcí, proto jej
buňka tak bedlivě hlídá.
Existuje
víc způsobů, jak udržovat vápníkovou rovnováhu. Nejčastěji je hladina
vápenatých iontů v buňce regulována tzv. vápníkovými pumpami, které jsou
umístěny v buněčné membráně a aktivně (buňku to stojí spoustu energie)
vápník odstraňují z buňky ven, nebo do vnitrobuněčných zásobáren (ve svalu
do tzv. sarkoplasmatického retikula) odkud může být použit k signalizaci
uvnitř buňky je-li třeba. Pak je několik enzymů, které jej specificky vyvazují,
a několik dalších cest.
V případě
kalpainu (kde se vápenaté ionty navazují na tzv. domému IV) dochází k jeho
stimulaci a tím zvýšení aktivity. A protože u typu dystrofie LGMD2A je navíc
kalpain defektní, nerozkládá sám sebe, začne se hromadit a ničit submembránový
skelet vlákna. Tak vznikají dystrofické
projevy. Příčinou tohoto typu dystrofie je tedy defekt v genu pro kalpain,
který sám sebe včas nezničí a začne devastovat vlákno. U DMD/BMD je to defekt
genu pro dystrofin, který se pak ve vlákně nevyskytuje nebo je defektní.
Vápenaté ionty přehnanou reakci kalpainu spouští a udržují v chodu, ale
samy o sobě nejsou příčinou tohoto typu dystrofie.
U všech
dystrofií je zvýšená hladina vápníku v buňce, protože membrána buňky je díky
nedostatku proteinů, které ji stabilizují, hodně křehká, má hodně
nefyziologických „děr“, nesedí v ní pořádně enzymové systémy (jako už
zmíněné pumpy) apod. Vápníku je pak v buňce příliš, aktivuje spoustu
enzymů, které začnou buňku sekundárně likvidovat. Pokud do buňky teče příliš
mnoho vápníku, nastává tzv. excitotoxická smrt, smrt z nadměrného
dráždění. Dystrofická svalová vlákna zvýšený tok vápníku v podstatě
“dorazí“, i když příčinou dystrofií sám o sobě není.
Jak to funguje?
Kalpain
inhibitor leupeptin je poměrně velká a nabitá molekula, která sama přes
membránu svalového vlákna neprojde. 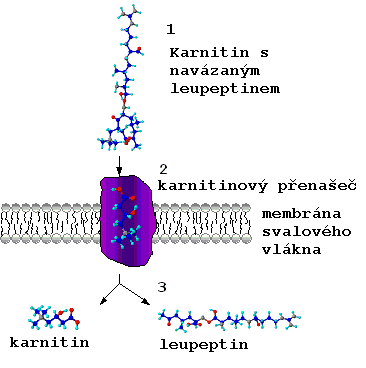 Potřebuje
k tomu pomocníka – molekulu karnitinu, látky, která je schopna leupeptin
navázat (1). Pro karnitin existují v membránách specifické přenašeče. Ty
jej rozpoznají, spolu s jeho „nákladem“ (leupeptinem) na sebe naváží a přenesou
dovnitř buňky (2). Ve svalovém vlákně se od sebe karnitin a leupeptin oddělí
(3) a leupeptin může plnit svoji úlohu – inhibici kalpainu 3.
Potřebuje
k tomu pomocníka – molekulu karnitinu, látky, která je schopna leupeptin
navázat (1). Pro karnitin existují v membránách specifické přenašeče. Ty
jej rozpoznají, spolu s jeho „nákladem“ (leupeptinem) na sebe naváží a přenesou
dovnitř buňky (2). Ve svalovém vlákně se od sebe karnitin a leupeptin oddělí
(3) a leupeptin může plnit svoji úlohu – inhibici kalpainu 3.
Leupeptin
inhibuje kalpain 3 tak, že se přes jednu ze svých nabitých skupin naváže na
aminokyselinový zbytek v jeho aktivním centru. Tím změní jeho prostorovou
strukturu a kalpain už nemůže štěpit bílkoviny napojené na membránu svalového
vlákna. Takto působí i spousta jiných látek, které inhibují enzymy.
Přínos
Myoduru je v tom, že obsahuje membránový přenašeč karnitin. Proto
specificky a efektivně zvyšuje přenos leupeptinu do srdeční a kosterní
svaloviny a brání její degradaci.
10.4.2004
·
Gene
therapy restores mouse lung function
·
Optimization of
power wheelchair control for patients with severe Duchenne muscular dystrophy
4.4.2004
·
Anti-TNFalpha
(Remicade) therapy protects dystrophic skeletal muscle from necrosis
·
New funding for Duchenne
muscular dystrophy research in UK
31.3.2004
Albuterol –
první studie na DMD a BMD pacientech
MDA podporovaná první studie studie
léku Albuterol pro DMD a mírnější variantu BMD provedená na UCLA (University of
California at Los Angeles ) ukázala nadějné výsledky.
Albuterol je lék patřící do
skupiny tzv. beta-2 agonistsů a je používán pro léčbu astma.
Již delší dobu se tušilo, že má pozitivní vliv na svalstvo, že zvyšuje tvorbu
svalových bílkovin a snižuje jejich rozklad. Toto potvrdila studie odbornice na
svaly Melisa Spencer a neurologa Michael Graves publikovaná v březnovém vydáni
Neurology.
Pokusu se zúčastnilo 9 chlapců ve věku od 5 do 9
lets DMD nebo BMD, všichni ještě chodící. Nejdříve byla změřena síla a
funkčnost svalů, potom byl chlapcům po dobu 12 týdnů podáván orálně albuterol
(Proventil Repitabs) nebo placebo.Ani děti ani lékaři netušili kdo kterou látku
užívá. Po 4 týdnech přerušení užívání léku pacienti, kteří dříve užívali
placebo, začali užívat albuterol a naopak, takže každý z nich užíval albuterol
buď v první nebo druhé části pokusu.
Když badatelé analyzovali výsledky pokusu zjistili,
že ti, kteří byli na albuterolu vykazovali větší svalovou výkonnost než ti, co
byli na placebu a to hlavně u steheních svalů. Ne u všech svalů došlo k
zesílení, badatelé se domnívají, že je to tím, že některé svaly absorbují
albuterol lépe než jiné. Funkční testy jako například doba chůze, běh na
vzdálenost 30 stop, vyjítí 4 schodů nebo zvednutí se z lehu neukázaly zlepšení.
Podle badatelů by léčba albuterolem musela pravděpodobně trvat déle, aby se to
projevilo na zlepšení funkčních testů. Žádné srdeční abnormality ( které byly předpokládány ) ani jiné
vedlejší účinky nebyly pozorovány.
Větší a pravděpodobně delší studie bude nezbytná k
tomu, aby potvrdila tyto výsledky a ujistila lékaře a rodiny, že tento lék je
bezpečný a účinný pro léčbu DMD a BMD.
30.3.2004
Další pokrok
týmu doktora Jacquese P. Tremblaye
Tým doktora Jacquese P. Tremblaye z Quebeku ohlásil
první úspěšný pokus na pacientech s DMD, jimž byly injikovány myoblasty
(nedospělé svalové buňky) od příbuzného dárce. Tyto myoblasty splynuly (sfúzovaly)
s nemocnými svalovými vlákny pacientů a indukovaly v nich expresi normální
genu pro dystrofin.
Do zatím první fáze déledobě plánovaného projektu
byli zahrnuti tři hoši ve věku 8, 10 a 16 let. Dva mladší byli ještě samostatně
pohybliví, nejstarší už byl upoután na vozík. Myoblasty dárců byly namnoženy
v tkáňové kultuře (laboratoř dr. Tremblaye je v Kanadě jediná, která
má povolení pro pěstování klinicky využitelných lidských buněk) a vpíchnuty
pacientům. Chlapci dostali sérii 25 injekcí, vpíchnutých těsně vedle sebe do
bérce (do svalu jménem musculs tibialis anterior). Každá injekce představovala
30 miliónů myoblastů vpravených do asi 1 centimetru krychlového.
Aby nedošlo k odvržení injikovaných myoblastů,
užívali pacienti relativně nové imunosupresivum (látku potlačující imunitní
reakci na tělu cizí buňky) Tacrolimus. Tacrolimus se ukázal jako nejvhodnější
imunosupresivum, neboť na rozdíl od např. cyklosporinu nebrání fúzi myoblastů a
neindukuje jejich apoptózu (programovanou buněčnou smrt) ještě před tím, než se
začnou v nemocné tkáni dělit. U ani jednoho z pacientů nebyly při
kontrolních odběrech prokázány protilátky proti buňkám dárců, u dvou starších
chlapců se vyskytovalo jen nemnoho lymfocytů v místě vpichů, u nejmladšího
žádné. Dva starší brali už před začátkem pokusu za účelem oslabení imunitních
reakcí Deflazacort, poslední žádné kortikoidy.
O měsíc později byly z míst, kam byly zdravé
myoblasty dárců injikovány, odebrány vzorky svalové tkáně (biopsie), které
umožnily zjistit výzkumníkům, zda podané myoblasty daly vzniknout svalovým
vláknům s dystrofinem. Dystrofin-pozitivních bylo v daných místech
6.8, 9.0 a 11.0% vláken, což je -vzhledem k ostatním dosud provedeným
klinickým studiím- značný úspěch. Tato dystrofin-pozitivní vlákna byly vlastně
podané buňky sfúzované s nemocnými vlákny. Dystrofin-poztivní byly i nové
buňky vzniklé fúzí myoblastů mezi sebou, ale ty nebyly do počtu
dystrofin-pozitivních vláken při hodnocení zahrnuty, neboť jejich fyziologický
význam je nejasný.
Počet vpíchnutých myoblastů, které přežijí alespoň
tři dny, není sice nijak vysoký, ale ty, co se v tkáni uchytí, se následně
dělí velmi úspěšně: 75-80% injikovaných myoblastů sice do tří dnů umře, ale ty,
co přežijí, je plně nahradí.
Dr. Tremblay říká: „Jsme těmito nadějnými výsledky
velmi potěšeni. Jako první na světě jsme uspěli vlastně s obnovením svalových
vláken produkujících dystrofin. První fáze našeho výzkumu otevírá slibné cesty
k potenciální léčbě.“
Další fází tohoto projektu bude injikovat zdravé
myoblasty do celého svalu, a to v počtu několika set injekcí. Jen tak bude
možno ověřit, zda tato technika může posílit celkovou sílu svalu, nebo alespoň
zabránit postupu jeho slábnutí. Skupina dr. Tremblaye nedávno od Health Canada
obdržela povolení zvýšit počet injekcí z 25 na 100.
Tato technika je nadějná nejen pro DMD pacienty, ale
i pro ty, co trpí recesivně zděděnou dystrofií. Na podobných technikách teď
pracuje i několik dalších týmů v USA a Francii. Časem by mohlo jít o
terapii vhodnou i pro poinfarktové stavy, kdy by bylo možno nahradit odumřelou
tkáň.
Další
podrobnosti : Dystrophin Expression
in Myofibers of Duchenne Muscular Dystrophy Patients Following Intramuscular
Injections of Normal Myogenic Cells.
29.3.2004
ADAM12 zmírňuje chorobné
změny kosterního svalstva u mdx myší
UM Wewer (1), B
Moghadaszadeh (1), R Albrechtsen (1), L Guo (2), M Zaik (3), N Kawaguchi (1),
RH Borup (4), P Kronqvist (1), HD Schroeder (5), KE Davies (6), R Hermann (3),
T Voit (3), FC Nielsen (4) and E Engvall (2). (1) Univ. of
Copenhagen, Copenhagen, Denmark; (2)The Burnham Inst., La Jolla, CA, U.S.A.;
(3) Univ. of Essen, Germany; (4) Copenhagen Univ. Hospital, Copenhagen,
Denmark; (5) Odense Univ., Odense, Denmark; (6) Univ. of Oxford, Oxford, U.K.
ADAM12 je disintegrin umístěný v membráně
buňky, v buňce má řadu funkcí. Vyskytuje se v
kosterním svalstvu během embrionálního vývoje a jeho tvorba ustává po narození.
U vyzrálých svalových buněk se objevuje opět při regeneraci. Vytvořili jsme
myši se zvýšenou tvorbou ADAM12 ve svalech. Po skřížení s mdx myšima jsme
dostali ADAM12/mdx myši, které se vyznačovaly výrazně nižšími chorobnými
změnami kosterního svalstva. ADAM12 chrání buňky před
odumřením, to bylo potvrzeno imunohistochemicky a sníženou hodnotou CPK. Dále
jsme dokázali, že ADAM12 zvyšuje tvorbu dvou komplexů : utrofin/DAG a alfa7-integrinu.
Toto je objev nového mechanismu kompenzace deficitu dystrofinu v kosterním
svalstvu. Předpokládáme, že ADAM12 je gen vylepšující fungování svalových buněk
a mohl by být využit k léčbě DMD.
24.3.2004
·
Erythropoietin: a new tool
for muscle disorders?
·
Rapid
identification of female carriers of DMD/BMD by quantitative real-time PCR
·
Management
of patients with Duchenne muscular dystrophy
·
Aquaporins
in skeletal muscle: reassessment of the functional role of aquaporin-4
·
Gene
therapy on muscular dystrophy
19.3.2004
·
Report on a Round Table
Conference in Monaco on 17 and 18 January 2004
·
New
findings on nerve cell proteins show promise for reducing disability
·
Advances
in AAV-mediated gene transfer for the treatment of inherited disorders
15.3.2004
Pro léčbu DMD Badatelé
kombinují kmenové buňky a genovou terapii
Louis Kunkel z Harvard Medical School a Jeffrey Chamberlain a Sheng Li z University of Washington oznámili , že kombinací terapií kmenovými buňkami a genovou terapií úspěšně léčí DMD myši. Myši byly léčeny speciálním typem svalových kmenových buněk označovaných SP (side population) do kterých byl přidán lidský gen pro dystrofin. SP buňky pochází z myších svalů a předpokládá se, že podobné buňky obsahují i lidské svaly. Badatelá injikovali SP buňky do ocasní žíly myší. Pak pozorovali jak buňky putují krevním oběhem a usazují se hlavně v poškozených oblastech svalů a produkují sice malé ale měřitelné množství lidského dystrofinu.
“Tato práce je jedním z
prvních kroků k léčbě neembrionickými kmenovými buňkami” tvrdí Kunkel a dodává,
že tím je vydlážděna cesta k pro použití pacientových geneticky opravených
kmenových buněk k léčbě. Badatelé věří, že tato technika minimalizuje riziko odmítnutí
kmenových buněk tělem pacienta.
13.3.2004
V dubnu se koná v San Francisku výroční setkání American Academy of Neurology. Je to pravděpodobně nejdůležitější celosvětové setkání neurologů. Na tomto setkáni bude prezentováno pouze 8 prací o DMD (o Parkinsonově chorobě 157, 257 o roztroušené skleróze). Příspěvky týkající se DMD :
·
Strength and Fatigue in mdx Mice Treated with
Weekly Oral Prednisolone for 52 Weeks
·
Creatine Monohydrate Supplementation Appears Safe
for Children with Neuromuscular Disorders
·
Combined Deficiency of Calpain and Dystrophin
Mutually Reduce the Severity of Phenotypes?
6.3.2004
· Specific cognitive deficits are common in children with Duchenne muscular dystrophy
· Readthrough of dystrophin stop codon mutations induced by aminoglycosides
· Steroid Treatment and the Development of Scoliosis in Males with Duchenne Muscular Dystrophy
2.3.2004
· ß2-Agonist administration reverses muscle wasting and improves muscle function in aged rats
29.2.2004
· Mini-dystrophin restores L-type calcium currents in skeletal muscle of transgenic mdx mice
· Transforming Growth Factor-ß1 Induces the Differentiation of Myogenic Cells into Fibrotic Cells in Injured Skeletal Muscle
· Dystrophin deletions and cognitive impairment in Duchenne/Becker muscular dystrophy
22.2.2004
21.2.2004
· Pharmaceutical patents : Medicament for treatment of Duchenne muscular dystrophy
14.2.2004
Výtah
z přednášky profesora Masafumi Matsuo z Kobe University Japonsko
Profesor Masafumo Matsuo referoval o svých experimentech s vystřižením exonu (exon skipping), které byly zaměřeny hlavně na korekci delece exonu 20 u pacienta s DMD. Když chybí exon 20 je exon 19 připojen přímo na exon 21. A protože exon 19 končí po prvním písmenu třípísmenného genetického slova je čtení exonu 21 posunuto, což vede k dekódování stop kodonu na začátku 21 exonu. Exon 19 začíná třípísmenným slovem. Jestliže jej tedy odejmeme, délka genu se zmenší o 110 bází z 3685. Tento o 3% kratší dystrofin by měl být částečně funkční, takže by pacient měl trpět daleko méně závažnou formou BMD.
Japonští
vědci experimentovali se sérií uměle vytvořených oligonukleotidů o délce 31
bází tvořených DNA a RNA vláknem. Z důvodu ochrany před buněčnými enzymy
byly chemicky modifikovány. Testovány byly na kultuře svalových buněk, myocytů,
pacienta s delecí 20 exonu. Až v 90% myocytů byl během pokusu detekován
dystrofin. Další oligonukleotidy byly vytvářeny i pro exony 41, 44, 45, 51 ,53
a 55, což jsou nejčastější poškozené exony.
Pro
první pokus nebyl použit oligonukleotid ale jednovláknová DNA s 31 sekvencemi.
Po sedmi dnech bylo možno ve vzorku svalové tkáně detekovat dystrofin, po 10
dnech bylo možno detekovat dystrofin ve 20% nedospělých svalových buněk.
Dalším
krokem bylo prokázat efektivitu u živých organizmů. Protože svalové
membrány mdx myší nemají dystrofin jsou pro potenciální lék podávaný do
krevního oběhu prostupnější než membrány u zdravých myší. Intraperitoneální
podání 2mg/kg váhy vedlo konečně k očekávanému vystřižení exonu 19. Po dvou
dnech byla detekována mRNA, a to nejen v kosterním svalstvu ale i v srdci a
vydržela tam nejméně 14 dní po léčbě.
Tato
in-vivo demonstrace otevřela cestu k prvnímu klinickému pokusu na člověku. Po
obdržení povolení od příslušné etické komise desetiletý chlapec s delecí exonu
20, který již musel používat vozík a jehož buňky byly použity k pokusům
in-vitro, obdržel 4 nitrožilní infůze a to jednu týdně po dobu 4 týdnů. Během
dvou hodin obdržel vždy 0,5mg DNA oligonukleotidu na 1kg váhy. Den po každé
infuzi byla dystrofinová mRNA detekována v lymfocytech pacienta. Její množství
rostlo po každé další infuzi. Tato nová RNA bylo o 0.64% menší než stále ještě
přítomná RNA s delecí 20 exonu. Týden po poslední infuzi byl pacientovi odebrán
vzorek svalové tkáně a imunologickými metodami konečně dokázán dystrofin. Další
a daleko detailnější analýzy této bílkoviny právě probíhají.
Zlepšení
svalové výkonnosti zatím nebylo detekováno, úroveň CK se také zatím výrazně nezměnila.
Oligonukleotidy, zdá se, jsou dostatečně stabilní protože byly detekovány ještě
několik týdnů po aplikaci. Pacient je nyní sledován a testován. Nyní je
plánováno opakování léčby čtyřmi následujícími infuzemi opět za rok. Možná ale
bude rozhodnuto opakovat infuze v měsíčních intervalech či dokonce 2x měsíčně.
Pokud
tato první klinická léčba dopadne tak úspěšně, jako to zatím vypadá, metoda
vystřižení exonu bude zkoušena i na chlapcích s delecemi exonu v oblasti 8-18,
20 –22, 20 –23 a i na rozsáhlejších delecích kdekoliv v oblsti exonu 20 –42.
Profesor Matsuo plánuje zahrnout do příštích klinických pokusů právě chlapce s
rozsáhlými delecemi v těchto oblastech.
Podle
posledních oficiálně ještě nepublikovaných informací prof. Matsuo předpokládá,
že dystrofin-pozitivní bude téměř 100% svalových vláken. Zdá se, že svalová výkonnost
pacienta se začíná lepšit, ikdyž je potřeba ještě další pozorování a testování,
aby to bylo možno potvrdit.
8.2.2004
25.1.2004 americká organizace Parent Project Muscular Dystrophy (PPMD) podepsala historickou
smlouvu s farmaceutickou korporací PTC
Therapeutics of
South Plainfield, New Jersey. Tato smlouva uvádí do pohybu výzkum Duchenneovy
svalové dystrofie. Jednání v oblasti výzkumu DMD PPMD iniciovala již v březnu
2003, Tento project, nazvaný Project Catalyst, je prvním oficiálním
partnerstvím PPMD s faramaceutickou firmou.
Vrámci projektu Catalyst provede PTC
Therapeutics pod vedením vědeckého ředitele PPMD Dr. Lee Sweeny rozsáhlý
automatizovaný průzkum s cílem objevit nový lék, který by omezil nebo dokonce
úplně zastavil degeneraci svalstva u pacientů s DMD. PPMD financuje tento výzkum
díky podpoře velkých sponzorů.
PPMD je největší organizace v USA zaměřená na Duchenneovu svalovou dystrofii. PPMD podporuje výzkum, vytváří standarty péče, snaží se urychlit vývoj léčby. Spojení usilí s biotechnologickým průmyslem je další z cest jak PPMD bojuje proti Duchenneově svalové dystrofii.
· Making
new muscle: Researchers in Rome produce a mouse that can regenerate its tissues
· Stem cell-mediated muscle regeneration is enhanced by local
isoform of insulin-like growth factor 1
· Extracellular
HMGB1, a signal of tissue damage, induces mesoangioblast migration and
proliferation
3.2.2004
Firma
Ceptor dceřinná společnost firmy Xechem
vyvinula lék MYODUR. Tento lék, jehož bezpečnost a účinnost již byla vyzkoušena
na zvířatech, obsahuje membránový transportér carnitin a dále leupeptin známý
jako calpain inhibitor. Calpain je primární proteaza, která vyvolává apoptozu
svalových buněk, u svalových dystrofií bývá v buňkách ve zvýšeném množství. Jak
carnitine tak leupeptin jsou pro FDA známe
látky, a protože vyrazně větší množství leupeptinu již bylo vyzkoušeno na
lidech Xechem věří, že nejde o rizikový vývojový program. Klinické pokusy by
měly začít v roce 2004 a protože jde o lék na těžkou a zatím neléčitelnou
chorobu bude FDA stačit
menši množství klinických testů. Samotné FDA je pak povinno prověřit podklady pro povolení léku
ve zkráceném termínu 6 měsíců Tento lék nevede k tvorbě plnohodnotného
dystrofinu, ale měl by zpomalit degradaci svalstva.
25.1.2004
· Outcome of non-invasive positive
pressure ventilation in paediatric neuromuscular disease
· Popping a pill could fix gene defect
20.1.2004
· Drugs May Help DMD-Affected Cells Read Through
Genetic Errors
Biofarmaceutická firma PTC Therapeutics postoupila
dále ve vývoji PTC124. Jde o lék,
který umožňuje buňkám obejít jisté typy genetických chyb. Není to ale všelék,
léčí jen jeden typ genetické poruchy – předčasný stop kodon. Nový lék by měl
fungovat s menší toxicitou než gentamicin, ale s podobným účinkem.
Studie na myších s DMD již prokázaly nižší toxicitu než u gentamicinu a
obnovu tvorby dystrofinu. PTC124 se zatím stále vyvíjí a testuje, klinické
pokusy se plánují na rok 2005.
· Supplement Test Results in
Duchenne MD Are Slightly Encouraging
· High-Dose Prednisone Trial in DMD
Revised, Now Open
17.1.2004
· Dystrophinopathy caused by mid-intronic substitutions activating cryptic exons in the DMD gene
· A-utrophin up-regulation in mdx skeletal muscle is independent of regeneration
16.1.2004
(
Článek z Molecular Therapy, který jsme avizovali 12.2.2003 v článku „Transplantace normálních a
geneticky pozměněných myoblastů“ )
11.1.2004
· Creatine
increases IGF-I and myogenic regulatory factor mRNA in C2C12 cells
9.1.2004
· Targeting the Immune System to
Improve Ventilatory Function in Muscular Dystrophy
5.1.2004
· Stem-cell
'secret of youth' found
2.1.2004
·
ViaCell, Amgen in cell therapies
deal
1.1.2004
· Myospryn is a novel binding partner for dysbindin in muscle.
·
Animal
models used to study Duchenne's muscular dystrophy
* Tyto stránky jsou určeny rodičům i lékařům.
Vše co považujeme za důležité pro rodiče se budeme snažit přeložit do
češtiny. Výrazy napsané kurzívou
nepřekládáme z důvodu nedostatečné znalosti odborné terminologie. Omlouváme se
za případné nepřesnosti při překladu a děkujeme za pochopení.