ABSTRACTS THAT WILL BE PRESENTED IN
THE 8TH ANNUAL MEETING OF THE AMERICAN SOCIETY OF GENE THERAPY, JUNE 1-5, ST
LOUIS, MISSOURI
1) Antisense Therapy for Duchenne Muscular
Dystrophy: A Realistic Possibility
Qi L. Lu, Adam Rabinowitz, HaiFang Yin, Julia
Alter, George Bou-Gharios, Terrence Partridge. Muscular Dystrophy Laboratory,
Neuromuscular/ALS Center, Carolinas Medical Center, Charlotte, NC; Muscle Cell
Biology, MRC Clinical Science Centre, London, United Kingdom; Department of
Renal Medicine, Imperial College, Hammersmith Hospital Campus, London, United
Kingdom
Duchenne Muscular Dystrophy (DMD) is the most
common form of muscular dystrophy affecting 1 in every 3500 live male births.
The disease is characterized by severe muscle wasting and weakness, which
becomes clinically evident between the ages of 3 to 5 years. The milder form of
the disease is Becker muscular dystrophy (BMD) with a spectrum of phenotypes
ranging from almost asymptomatic to mild forms of DMD. Both DMD and BMD are the
results of mutations in the dystrophin gene. The phenotypic distinction between
DMD/BMD can be explained by the reading-frame theory where DMD is caused by
frame-shifting or nonsense mutations leading to premature termination of protein
synthesis whereas BMD is caused by mutations resulting in altered, but in-frame
transcripts. One promising alternative approach for treatment of DMD is
antisense oligonucleotide (AO)-mediated gene correction at RNA level (antisense
therapy). This technique uses small oligonucleotides to correct frame-shifting
or nonsense mutations by skipping a mutated exon or exons which disrupt the
reading frame in such a way that restores reading frame of the dystrophin
transcript. This results in the expression of a truncated, but at least
partially functional dystrophin. Antisense therapy is uniquely applicable to DMD
treatment for many reasons. First, the gene consists of 79 exons and muscle form
of dystrophin protein can be divided into amino terminal, rod, cysteine-rich and
carboxy terminal domains. However, the rod domain of the dystrophin gene appears
not to be critical for its functions. Second, the majority of DMD mutations
occur within this non-critical region of the dystrophin gene. Thus correction of
frame-shifting mutations by skipping the mutated exon or other exons necessary
for restoration of reading frame will retain critical functions of the protein.
We have demonstrated that specifically designed 2 -O-methyl
phosphorothioate AO (20MeAO) delivered by intramuscular injections was able to
effectively skip the mutated region (exon) of the dystrophin gene in animal
model of DMD with functional improvement in the muscles. Dystrophin induced by
20MeAO remains at detectable levels even 3 months after intramuscular
injections. This instigated clinical trials in several countries. We now
demonstrated that the AOs delivered by simple intra-venous injections can induce
dystrophin expression in body-wide skeletal muscles up to therapeutic levels.
The simplicity and safety of the antisense therapy provide a realistic
possibility for treatment of the majority of DMD mutations.
-O-methyl
phosphorothioate AO (20MeAO) delivered by intramuscular injections was able to
effectively skip the mutated region (exon) of the dystrophin gene in animal
model of DMD with functional improvement in the muscles. Dystrophin induced by
20MeAO remains at detectable levels even 3 months after intramuscular
injections. This instigated clinical trials in several countries. We now
demonstrated that the AOs delivered by simple intra-venous injections can induce
dystrophin expression in body-wide skeletal muscles up to therapeutic levels.
The simplicity and safety of the antisense therapy provide a realistic
possibility for treatment of the majority of DMD mutations.
2) Ex Vivo Gene Therapy for Duchenne
Muscular Dystrophy: Lentiviral and PhiC31 Integrase
Approaches
Simon P.
Quenneville, Pierre Chapdelaine, Joël Rousseau, Michele P. Calos, Jacques P.
Tremblay. Genetique Humaine, CRCHUL, Sainte-Foy, QC, Canada; Department of
Genetics, Stanford University School of Medecine, Stanford,
CA
Duchenne muscular dystrophy is the most severe muscular
dystrophy. It is caused by the absence of dystrophin in muscle fibers. This
absence lead to increased muscle damage, loss of muscle mass, loss of strength,
respiratory and heart failure. This disease as currently no treatment. Myogenic
cells transplantation is a possible cure for Duchenne muscular dystrophy.
However, allogeneic graft success relies on the use of efficient but toxic
immunosuppressive drugs. The use of these drugs is a major problem that could be
solved by the use of ex vivo gene therapy. This method consists in genetically
modifying patient myoblasts before their auto-transplantation. In this study, a
viral and a non viral method were tested to perform the genetic modification.
The co-transfection (nucleofection) of a PhiC31 integrase and a dystrophin
expressing plasmid as already led to a stable expression of the full length
dystrophin1. This was the biggest expression cassette ever stabilized
in primary cultured human myoblasts. Using this method, we are now capable of
transferring transgene expressing cells into a muscle, leading to expression
into the muscle fibers. However, this method is not very efficient. This problem
could be solved using viral vectors. We have generated eGFP and
eGFP-micro-dystrophin expressing lentiviral vectors under the control of the CMV
and MCK promoters. Following in vitro infection of human myoblasts, the cells
were engrafted into SCID mice muscles. We were able to detect the expression of
both transgenes into these muscle fibers one month after the engraftment. Evans
bleu will be injected in the blood of the mice before an eccentric exercise. We
will verify whether the expression of dystrophin in the muscle fibers reduces
their damage during the exercise and thus the incorporation of Evans blue. This
work indicates that ex vivo gene therapy is a possible approach to treat
Duchenne muscular dystrophy.
1-Quenneville SP et al., Mol Ther. 2004
Oct;10(4):679-87.
3) Immunity to AAV-Mediated Gene Therapy in a
Random-Bred Canine Model of Duchenne Muscular Dystrophy
Zejing Wang, Michael J. Blankinship, Paul
Gregorevic, Marie-Terese Little, Rainer J. Storb, James M. Allen, Stephen J.
Tapscott, Jeffrey S. Chamberlain, Christian S. Kuhr. Transplantation Biology,
Fred Hutchinson Cancer Research Center, Seattle, WA; Human Biology, Fred
Hutchinson Cancer Research Center, Seattle, WA; Neurology, University of
Washington, Seattle, WA
Introduction: Duchenne
muscular dystrophy (DMD) is caused by mutations in the dystrophin gene. Studies
in the mdx mouse model of DMD have shown that muscle membrane integrity and
function can be improved by AAV-mediated delivery of a functional dystrophin
protein. To assess the potential clinical utility of treating human DMD patients
with AAV-mediated gene delivery, we performed a series of direct intra-muscular
injections in random-bred normal dogs and in dogs with muscular dystrophy caused
by a dystrophin mutation (xmd dogs). Methods: AAV serotypes 2 and 6
carrying different promoter-transgene cassettes were produced as previously
described for the murine studies and purified either on a heparin column or with
a combination of heparin column and cesium chloride gradient. Direct
intramuscular injections of virus (1012, 1010, or
108 viral genomes per site) in a total volume of 250 ul were
performed. The injection sites were biopsied under anesthesia between 2 and 12
weeks after injection. Results: Injection of either AAV6 or AAV2
expressing CMV-b-galactosidase (b-gal) induced a strong inflammatory response
containing both CD8+ and CD4+ T-lymphocytes with peak tissue destruction 4 weeks
following viral injection. A similar robust immune response was seen with
injection of AAV6-RSV-alkaline-phosphatase, AAV6-CMV-canine-factor-IX (cFIX) and
with empty AAV6 capsid alone. Additional purification of the AAV6-CMV-cFIX by
centrifugation through a cesium chloride gradient did not diminish the immune
response. Continuous immunosuppression with cyclosporine (CSP) and mycophenolate
mofetil (MMF) largely prevented the immune response to AAV6-CMV-b-gal in a
normal dog for up to four weeks and permitted robust transgene expression. The
same immunosuppressive regimen did not prevent an immune response to
AAV6-CMV-cFIX or AAV6-CMV-human-micro-dystrophin in an xmd dog, suggesting that
a more aggressive immunosuppressive regimen might be necessary in the xmd dog.
Conclusions: Taken together, our results suggest that AAV capsid
proteins, or proteins associated with the capsid, elicit significant immune
responses when directly injected into skeletal muscle of normal random-bred
dogs. The combination of CSP and MMF effectively prevents the immune responses
in a normal dog, but not in an xmd dog, possibly due to the pre-existing
inflammatory nature of the DMD muscle disease or due to genetic variation in a
random-bred animal population. The robust immune responses to AAV2 and AAV6 in
random-bred dogs contrast with the lack of an immune response in studies by
others in inbred mice and dogs. Further studies will be necessary to determine
the nature of the immune responses and to develop appropriate immunosuppressive
regimen for AAV gene delivery to xmd muscle.
4) Stable Genome Alteration of the Dystrophin
Gene for the Treatment of Duchenne Muscular Dystrophy (DMD) Due to Frame-Shift
Mutations Using Oligonucleotide-Mediated Gene Editing
Carmen Bertoni, Thomas A. Rando. Neurology,
Stanford Universdity School of Medicine, Stanford,
CA
Duchenne muscular dystrophy is an X linked neuromuscular
disorder characterized by lack of dystrophin gene expression. Mutations in the
dystrophin gene include single point mutations, large deletions and frame shift
mutations. Genome editing of the dystrophin gene represents an attractive
approach to restoration of dystrophin into skeletal muscle of DMD patients.
Oligonucleotide-mediated single base substitution has the potential to
completely correct the mutation in the case of single point mutations, or to
restore the reading frame of the dystrophin gene to correct frame shift
mutations. Furthermore oligonucleotide-mediated gene correction allows the gene
to remain under the control of its own regulatory mechanisms.
We have tested
the applicability of genome editing of the dystrophin gene in the mdx
mouse model for DMD. This strain contains a stop codon in exon 23 of the
dystrophin gene that is responsible for the absence of dystrophin protein in
skeletal muscles. As a target for the single base substitution we have chosen
the splice boundary of exon 23 of the mouse dystrophin gene in order to induced
exon skipping to bypass the nonsense mutation and induce expression of
internally deleted but functional dystrophin proteins. We have designed single
stranded oligonucleotides (ssODNs) complimentary to the coding or the non-coding
strand of the donor site of exon 23. Modifications were inserted into the
oligonucleotides in an attempt to recruit specific repair processes present in
eukaryotic cells and increase the efficiency of the repair process. For these
kinds of studies, muscle cells represent an very appealing system since the
repair processes involved can be characterized in dividing cells (proliferating
muscle precursor cells) as well as mature myofibers at the terminally
differentiated stage, thus providing further insight in the process that
regulate gene correction.
CpG modifications were introduced at the 3 end
of ssODNs as well as at the targeted base. The ability of these modified ssODNs
to increase gene repair was studied in muscle cells both in vitro and
in vivo. Correction of the dystrophin gene was shown to occur at the
genomic level using all types of oligonucleotides and was shown to be stable
over prolonged periods of time. In muscle cells in culture, restoration of
dystrophin expression was analyzed at the protein level by western blot and
immunostaining analysis, and at the mRNA level by RT-PCR. In vivo
analysis also showed restoration of dystrophin in skeletal muscles of injected
mice by immunostaining. These studies confirm the applicability of genome
editing in cases in which DMD is due to single point and frame-shift
mutations.
5) Embryonic Stem Cell-Mediated Regenerative
Therapy for Duchenne Muscular Dystrophy
Shiro Ozasa, Shigemi Kimura, Makoto Matsukura,
Kaori Ito, Makoto Ikezawa, Hiroe Kawano, Teruhisa Miike, Kenichi Yamamura, Kimi
Araki, Kuniya Abe Hitoshi Niwa. Department of Child Development, Kumamoto
University Graduate School, Kumamoto, Japan; Institute of Molecular Embryology
and Genetics, Kumamoto University Graduate School, Kumamoto, Japan; Research
& Development Team for Mammalian Cellular Dynamics, BioResource Center,
RIKEN Tsukuba Institute, Tsukuba, Ibaragi, Japan; Laboratory of Pluripotent Cell
Studies, RIKEN center for developmental Biology, Kobe, Hyogo,
Japan
Introduction
Duchenne muscular dystrophy (DMD) is
a severe muscle degenerative disorder caused by mutations in the dystrophin
gene. Embryonic stem cell (ES cell) transplantation is one of the more promising
therapies because ES cells are available in large quantities and can serve to
systemically restore affected muscles of DMD patients. Although ES cells have
shown to be capable of differentiating into various tissues and cell types, it
has been difficult to induce differentiation of ES cells specifically into
muscle fibers only. We are proposing a method to overcome this impediment by
establishing genetically engineered ES cells which harbor a
tetracycline-regulated expression vector encoding the myogenic transcriptional
factor MyoD.
Methods and Results
ZHTc6 is a mouse-derived ES cell line in
which Tet-Off System is integrated. ZHTc6 can be induced to express the POU
transcriptional factor Oct-3/4 on the removal of doxycycline, a derivative of
tetracycline. We cultured ZHTc6 without feeders in LIF-supplemented medium. We
constructed Supertargeting Vector-MyoD, which is targeted upon the
electroporation to invoke the homologous recombination and replace the inducible
Oct-3/4 gene of ZHTc6 with MyoD gene. Southern-blot analysis revealed that three
out of 6 clones were recombinants. The recombinants, hereinafter referred to as
ZHTc6-MyoDs, were induced to differentiate nonspecifically in the
differentiation medium in the absence of doxycycline. Western analysis of the
differentiated cells revealed the expression of MyoD, desmin, myogenin and fast
myosin heavy chain (fMHC). The involvement of MyoD in myotube formation was
substantiated by colocalization of MyoD with the muscle-specific protein
dystrophin as revealed by immunocytochemical analysis. Desmin and fMHC were also
detected by immunocytochemical analysis. Serial flow cytometric analysis
revealed that the undifferentiated ZHTc6-MyoDs initially expressed Sca1 and
c-kit at a high level (more than 90%), but the expression gradually decreased
during differentiation. To the contrary, only a small fraction (less than 1%) of
ZHTc6-MyoDs expressed CD34 throughout the differentiation. We intramuscularly
injected 2-day-differentiated ZHTc6-MyoDs into gastrocnemius muscles of adult
mdx-nude mice. Seven weeks after injection, tumors developed in the injected
area to the size of 20 mm in diameter. Examination of the tumor specimen
uncovered the presence of the clusters of dystrophin-positive myofibers
surrounded by granular tumor cells.
Conclusion
In this system, we can
maintain ES cells undifferentiated in the presence of doxycycline and induce ES
cells to undergo myogenic specification on the removal of doxycycline in
vitro.
6) Development of rAAV-Based Skeletal and
Cardiac Muscle Regulatory Cassettes for Gene Therapy of Duchenne Muscular
Dystrophy
Maja Z. Salva,
Charis L. Himeda, Phillip Tai, Michael J. Blankinship, Paul Gregorevic, James M.
Allen, Leonard A. Meuse, Jeffrey S. Chamberlain, Stephen D. Hauschka.
Bioengineering, Univeristy of Washington, Seattle, WA; Biochemistry, University
of Washington, Seattle, WA; Neurology and Senator Paul D. Wellstone Muscular
Dystrophy Cooperative Research Center, University of Washington, Seattle,
WA
A promising gene therapy approach for treatment of DMD
involves systemic delivery of rAAV vectors encoding microdystrophin. This method
was shown to mediate efficient transduction of all striated muscles and
high-level transgene expression driven by the ubiquitous CMV promoter. However,
the use of viral promoters increases the risk of an immune response, largely due
to transgene expression in antigen-presenting cells, and may thus be inadequate
for long-term therapy. An ideal regulatory cassette for gene therapy of DMD thus
needs to direct high-level, tissue-specific expression in skeletal and cardiac
muscle and be short enough to package into the rAAV microdystrophin
construct.
Our lab has developed a series of muscle-specific regulatory
cassettes based on the murine muscle creatine kinase (MCK) gene enhancer and
promoter regions. The previous 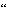 best
best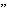 small cassette, CK6 (580bp) drives microdystrophin expression in skeletal muscle
following systemic rAAV delivery, at 10% of the level of the CMV promoter.
However, expression in heart and diaphragm is very low. To improve expression,
we combined the wild type MCK enhancer with a 190bp enhancer region from the
murine alpha myosin heavy chain (MyHC) gene, which is expressed at high levels
in cardiac muscle. Addition of the MyHC enhancer increased activity in MM14
skeletal myocytes by 5-fold as assessed by alkaline phosphatase reporter gene
activity. Furthermore, high-level expression was detected in skeletal muscle, as
well as heart and diaphragm after systemic delivery in mice of rAAV6 vectors
encoding these constructs. The activity of the hybrid cassette was 7-fold higher
in heart and 3-fold higher in diaphragm and soleus (predominantly slow-twitch
muscle fibers) when compared to the wild type MCK cassette, while the activity
in tibialis anterioris (predominantly fast-twitch muscle fibers) was unchanged.
Expression was restricted to muscle tissue, as evidenced by nearly undetectable
levels in the liver, spleen, lung, and aorta. Additional cassettes comprising
the MyHC enhancer and various modifications of the minimal MCK cassette have
also been tested in MM14 skeletal myocytes. The strongest cassettes were shown
to possess 12-fold higher activity than the wild type MCK cassette, and were
about 55% as active as the CMV promoter. These contained a 63bp deletion within
the MCK enhancer, addition of a 50bp region downstream of the transcription
initiation site, and a mutation that creates a consensus initiator-binding site
from the terminal transferase promoter. We are currently testing activity of
these constructs in mouse muscle and non-muscle tissues following systemic
delivery of rAAV6 vectors. The strongest tissue-specific cassette will be
evaluated for long-term expression of therapeutic levels of microdystrophin in
the mdx mouse model for DMD.
small cassette, CK6 (580bp) drives microdystrophin expression in skeletal muscle
following systemic rAAV delivery, at 10% of the level of the CMV promoter.
However, expression in heart and diaphragm is very low. To improve expression,
we combined the wild type MCK enhancer with a 190bp enhancer region from the
murine alpha myosin heavy chain (MyHC) gene, which is expressed at high levels
in cardiac muscle. Addition of the MyHC enhancer increased activity in MM14
skeletal myocytes by 5-fold as assessed by alkaline phosphatase reporter gene
activity. Furthermore, high-level expression was detected in skeletal muscle, as
well as heart and diaphragm after systemic delivery in mice of rAAV6 vectors
encoding these constructs. The activity of the hybrid cassette was 7-fold higher
in heart and 3-fold higher in diaphragm and soleus (predominantly slow-twitch
muscle fibers) when compared to the wild type MCK cassette, while the activity
in tibialis anterioris (predominantly fast-twitch muscle fibers) was unchanged.
Expression was restricted to muscle tissue, as evidenced by nearly undetectable
levels in the liver, spleen, lung, and aorta. Additional cassettes comprising
the MyHC enhancer and various modifications of the minimal MCK cassette have
also been tested in MM14 skeletal myocytes. The strongest cassettes were shown
to possess 12-fold higher activity than the wild type MCK cassette, and were
about 55% as active as the CMV promoter. These contained a 63bp deletion within
the MCK enhancer, addition of a 50bp region downstream of the transcription
initiation site, and a mutation that creates a consensus initiator-binding site
from the terminal transferase promoter. We are currently testing activity of
these constructs in mouse muscle and non-muscle tissues following systemic
delivery of rAAV6 vectors. The strongest tissue-specific cassette will be
evaluated for long-term expression of therapeutic levels of microdystrophin in
the mdx mouse model for DMD.
7) Enhanced Plasmid-Mediated Dystrophin
Expression in the mdx Mouse Model for Duchenne Muscular Dystrophy by a
PhiC31 Integrase Plasmid System
Carmen Bertoni, Sohail Jarrahian, Thurman M.
Wheeler, Yining Li, Eric C. Olivares, Michele P. Calos, Thomas A. Rando.
Neurology, Stanford University School of Medicine, Stanford, CA; Genetics,
Stanford University School of Medicine, Stanford,
CA
Duchenne muscular dystrophy is caused by lack of
dystrophin expression in skeletal muscles and is characterized by progressive
degeneration of muscle fibers. To be effective, gene therapy approaches to DMD
need to target a large number of fibers in the muscle, and the distribution of
dystrophin through the fiber length needs to be sufficient to prevent fiber
degeneration. Plasmid-based gene therapies have been shown to be a valid
approach to the treatment of a variety of disorders including DMD. Concerns,
however, have been raised about the ability of extrachromosomal vectors to
sustain gene expression for prolonged periods of time at levels that are
therapeutic.
We have tested the ability of the phiC31 integrase system to
integrate plasmid vectors stably into the chromosomes of terminally
differentiated myonuclei and maintain transgene expression over prolonged
periods of time. Tibialis anterior muscles of wild-type animals were injected
with a plasmid carrying a luciferase reporter gene under the control of the CK6
muscle specific promoter and a phiC31 integrase attB site. One group of
muscles was co-injected with an equal amount of a CMV-driven
integrase-expressing plasmid (pCSI) to direct site-specific integration, while
contralateral muscles received empty vector (pCS). All muscles were subjected to
electroporation to achieve a high level of plasmid delivery, and expression was
followed using a bioluminescence live imaging system (BLIS). Shortly after
delivery, the level of gene expression obtained in muscles that had received
pCSI was 10-fold higher than in muscles receiving pCS. This increase in the gene
expression level was maintained for up to 2 years after injection. Site-specific
integration of the luciferase attB plasmid was confirmed by PCR
analysis.
The beneficial effects of phiC31-mediated integration were also
tested in the mdx mouse model for DMD. Muscles were co-injected with an
attB plasmid carrying the luciferase reporter gene driven by the CK6
promoter and an attB plasmid carrying the full-length dystrophin gene,
also driven by the CK6 promoter, in the presence of either pCS or pCSI. BLIS
analysis showed persistent luciferase expression only in muscles co-injected
with the integrase expression vector. The level of dystrophin expression was
higher in muscles co-injected with pCSI, as demonstrated by RT-PCR analysis. Six
months after gene delivery, the overall increase in dystrophin expression
resulted in more than twice as many dystrophin-positive fibers in pCSI-injected
muscles, compared to muscles injected with pCS. Distribution of dystrophin along
the length of the fibers was also greater in muscle receiving the integrase
plasmid. Plasmid integration was also confirmed in muscles co-injected with
pCSI. CD4 and CD8 staining showed no increase in the inflammatory response in
muscles that received integrase. Site-specific integration of plasmid DNA
mediated by phiC31 integrase enhance plasmid-mediated gene expression and has a
relevant clinical application for muscle diseases such as
DMD.
8) Sustained Whole-Body Functional Rescue by
Systemic Delivery of AAV8 Vectors in Heart Failure and Muscular Dystrophy
Hamsters
Tong Zhu, Liqiao
Zhou, Satsuki Mori, Zhong Wang, Charles Mctiernan, Chunping Qiao, Chunlian Chen,
Daowen Wang, Juan Li, Xiao Xiao. Molecular Genetics and Biochemistry, University
of Pittsburgh School of Medicine, Pittsburgh, PA; Cardiovascular Institute,
University of Pittsburgh School of Medicine, Pittsburgh, PA; Cardiology, Tongji
Hospital,Huazhong Science & Technology University, Wuhan, Hubei,
China
The success of muscular dystrophy gene therapy
requires widespread and stable gene delivery with minimal invasiveness. Using
the delta-sarcoglycan (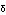 SG)-deficient
hamster (TO-2), a congestive heart failure and muscular dystrophy hamster model,
here we show that a single injection of double-stranded adeno-associated virus
serotype 8 vector carrying human delta-sarcoglycan gene (dsAAV8-SG), without
the need of any physical or pharmaceutical interventions, achieved nearly
complete gene transfer and tissue-specific expression in the heart and skeletal
muscles of the diseases hamsters.
SG)-deficient
hamster (TO-2), a congestive heart failure and muscular dystrophy hamster model,
here we show that a single injection of double-stranded adeno-associated virus
serotype 8 vector carrying human delta-sarcoglycan gene (dsAAV8-SG), without
the need of any physical or pharmaceutical interventions, achieved nearly
complete gene transfer and tissue-specific expression in the heart and skeletal
muscles of the diseases hamsters.
Broad and sustained ( >12 months)
restoration of the missing delta-sarcoglycan gene in the TO-2 hamsters corrected
muscle cell membrane leakiness throughout the body, and normalized serum
creatine kinase levels (an 50-100 fold drop). Histology examination revealed
minimal or absence of central nucleation, fibrosis, and calcification in the
skeletal muscle as well as the heart. Whole body functional analysis such as
treadmill running showed dramatic improvement, similar to the wild-type F1B
hamsters. Furthermore, cardiac function studies such as echocardiography
revealed significantly increased %FS and decreased LVEDD and LVESD in the
treated TO-2 hamsters. The survival time of treated TO-2 hamsters was
dramatically prolonged.
To the summary, our study demonstrates that systemic
AAV8 vector-mediated gene transfer could persistently ameliorate of cardiac and
skeletal muscle pathology, profoundly improve cardiac and whole-body functions,
and significantly prolong the life-span of the treated TO-2 hamsters. Future
research is warranted to see if such profound gene delivery efficiency as well
as therapeutic efficacy can be duplicated in large animals and eventually in
human patients.
9) Dystrophin in Vascular Smooth Muscle Is
Important for Duchenne Muscular Dystrophy Therapy
Kaori Ito, Shigemi Kimura, Gail D. Thomas, Shiro
Ozasa, Makoto Matsukura, Makoto Ikezawa, Hiroe Ueno, Kowashi Yoshioka, Misao
Suzuki, Takeshi Miwa Teruhisa Miike. Department of Child Development, Faculty of
Medical and Pharmaceutical Sciences Kumamoto University Graduate School,
Kumamoto, Japan; Department of Internal Medicine, Division of Hypertension,
University of Texas Southwestern Medical Center, Dallas, TX; Division of
Transgenic Technology Center for Animal Resources and Development (CARD),
Kumamoto University, Kumamoto, Japan; Department of Oncogene Research, Research
Institute for Microbial Deseases, Osaka University, Suita, Osaka,
Japan
Duchenne muscular dystrophy (DMD) is an X-linked
fatal disease caused by mutations of the gene encoding the cytoskeletal protein
dystrophin, which is associated with a complex of proteins that spans the
membrane and effectively links the cytoskeleton to the extracellular matrix.
Despite the abundance of information about the molecular basis of this disease,
there is currently no effective treatment for DMD because the mechanism by which
dystrophin deficiency produces the clinical phenotype is poorly understood. Our
previous data showed that dystrophin expression is normally detectable in smooth
muscle cells (SMCs) as well as in skeletal muscle cells. Likewise, it is
defective in SMCs of DMD patients and mdx mice, an animal model of DMD. Recent
studies have revealed that focal multicellular myocytolytic lesions in muscular
dystrophies may represent functional vascular abnormalities, regardless of
whether or not they are primary. Although vascular dysfunction in DMD patients
and mdx mice has been also reported, it is considered that the dystrophin
deficiency of skeletal muscle and/or vascular endothelium, not vascular smooth
muscle cells (VSMCs), is responsible for the vascular abnormalities.
We have
hypothesized that dystrophin deficiency of VSMCs leads to vascular dysfunction
and exacerbates muscle pathology. Here we generated transgenic mice
expressing14Kb full-length human dystrophin cDNA under the transcriptional
control of the smooth muscle alpha-actin promoter. These mice were then crossed
with mdx mice, resulting in three independent SMTg/mdx lines, that express
dystrophin only in SMCs. The expression pattern was detectable by
semi-quantitative RT-PCR analysis and immunohistochemical staining, which showed
the specific expression of transgene in SMCs.
We assessed the
vasoconstrictor response to sympathetic stimuli to determine if dystrophin
expression of VSMCs affects the blood flow regulation during exercise. In
contrast to mdx mice, alpha-adrenergic vasoconstriction in SMTg/mdx mice is
significantly attenuated during muscle contraction, resulting in amelioration of
the regulation of blood flow. Furthermore serum CK levels of SMTg/mdx mice were
significantly reduced in comparison with those of mdx mice. This is the first
report of a functional role for dystrophin in VSMCs and the resulting partial
restoration of phenotype. These results suggest that VSMCs can be an effective
target for treatment of DMD. Further, we believe that our SMTg/mdx mouse model
is worth exploring to gain a better understanding of the functions of dystrophin
in VSMCs and the pathophysiology of DMD patients.
10) An Ideal Therapeutic Agent for Duchenne
Muscular Dystrophy Involving a Gutted Adenovirus Expressing Full-Length
Utrophin
Jatinderpal R.
Deol, Renald Gilbert, Mylene Bourget, Joon-Shik Moon, Josephine Nalbantoglu,
George Karpati. Neuromuscular Research, Montreal Neurological Institute,
Montreal, QC, Canada; Genomics and Gene Therapy Vectors, Biotechnology Research
Institute, Montreal, QC, Canada; Department of Neurology, UMDNJ-New Jersey
Medical School, Newark, NJ
Duchenne muscular dystrophy
(DMD) is characterized by necrosis and progressive loss of muscle fibers. DMD
patients have a mutation in the gene encoding dystrophin (dys), a large
sarcolemmal membrane-associated cytoskeletal protein. Gene replacement therapy
using fully deleted adenoviral vectors shows great potential for the eventual
treatment of DMD.These vectors are less immunogenic than their predecessors and
have the capacity to carry the full-length dys cDNA (13 kb). The introduction of
fully deleted adenoviral (AdV) vectors encoding full-length dys into muscles
leads to significant improvement of the dystrophic phenotype of the mdx
mouse, an animal model of DMD. However, the introduction of dys, a neoantigen,
into mdx muscle causes immune responses resulting in increased appearance
of inflammatory infiltrates and damage to the muscle. An alternative approach is
to use utrophin (utr), a functional homologue of dys, normally present only at
the neuromuscular junction. This approach of simply increasing the expression of
a protein that is already present in dystrophic muscle would prevent any immune
response to the transgene product. Thus, we have created a fully deleted AdV
encoding full-length murine utr cDNA regulated by a powerful hybrid promoter
consisting of the chicken b-actin promoter and CMV enhancer. Cells infected with
the purified recombinant AdV showed strong utr expression levels as determined
by Western blot analysis. Furthermore, during in vivo studies, high
transduction levels were obtained in the tibialis anterior (TA) muscles of
mdx mice. The vector was administered to cohorts of neonates (2-4 day
old) and adults (5 to 7 week old) at doses of 1.45 x 1010 and 4.35 x
1010 virus particles respectively. In the first group of neonates
(n=5) at 10 days post-injection the mean number of sarcolemmal utr-positive
fibers in injected vs control-injected TA was 1596 +/- 297 vs 114 +/- 76, which
corresponds to 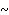 58 % transduction level. The mean number of utr-positive fibers
in the adults (n=7) was 685 +/- 505 and 112 +/- 80 for viral and control
injected TAs respectively, which corresponds to 23 % transduction
level. The numbers obtained correspond to an approximate 14 and 6 fold increase
in utr expression over control levels in neonates and adults. These results were
further validated by consistent utr levels observed by Western blots using
muscle sections. Subsequent in vivo evaluations will be performed at 30,
60, 90, 180, and 360 days post-injection. The injected muscles will be examined
for utr expression, the restoration of the dys-associated protein complex, and
the reversal of the physiological dystrophic phenotype. Considering our
preliminary data thus far, helper-dependent, fully deleted (gutted) AdV
expressing full length utr promises to be the ideal agent for gene replacement
therapy in dys-deficiency states.
58 % transduction level. The mean number of utr-positive fibers
in the adults (n=7) was 685 +/- 505 and 112 +/- 80 for viral and control
injected TAs respectively, which corresponds to 23 % transduction
level. The numbers obtained correspond to an approximate 14 and 6 fold increase
in utr expression over control levels in neonates and adults. These results were
further validated by consistent utr levels observed by Western blots using
muscle sections. Subsequent in vivo evaluations will be performed at 30,
60, 90, 180, and 360 days post-injection. The injected muscles will be examined
for utr expression, the restoration of the dys-associated protein complex, and
the reversal of the physiological dystrophic phenotype. Considering our
preliminary data thus far, helper-dependent, fully deleted (gutted) AdV
expressing full length utr promises to be the ideal agent for gene replacement
therapy in dys-deficiency states.
11) A Novel Approach of Gentamicin Therapy
for Duchenne Muscular Dystrophy Using Hybrid Liposome and Establishment of a
System To Identify the Patients Eligible for the
Treatment
Shigemi Kimura,
Kaori Ito, Shro Ozasa, Hiroe Kawano, Makoto Ikezawa, Makoto Matsukura, Ryuichi
Ueoka, Yoko Matsumoto, Msafumi Matsuo, Yasuhiro Takeshima Teruhisa Miike.
Department of Child Development, Kumamoto University Graduate School, Kumamoto,
Japan; Department of Applied Life Science, Sojo University, Kumamoto, Japan;
Department of Pediatrics, Kobe University Graduate School of Medicine, Kobe,
Japan
Aminoglycoside antibiotics have been found to
suppress nonsense mutations located in the defective dystrophin gene in mdx
mice, suggesting a possible treatment for Duchenne muscular dystrophy (DMD).
However, the gentamicin therapy is hindered by several problems. First, a high
dose of gentamicin, which is needed to induce a read-through of the nonsense
mutations in DMD patients, can not be given because of oto- and nephrotoxicity.
Even if this dose could be tolerated, this high concentration cannot be
sustained due to the short half-life of gentamicin. Lastly, identifying patients
that are applicable for this therapy is difficult because: 1) only 5 to 13% of
DMD patients have nonsense mutations in the dystrophin gene, 2) it is
challenging to find nonsense mutations in the gene because dystrophin cDNA is
very long (14kb), and 3) the efficiency of aminoglycoside-induced read-through
is dependent on the kind of nonsense mutation.
In order to overcome the dose
and concentration issues, we have investigated the use of hybrid liposomes. Mdx
mice, given hybrid liposome encapsulated gentamicin (340mg/kg) intraperitoneally
for 2 weeks, did not show oto- and nephrotoxicity. In contrast, only mdx mice
injected with the same dose of gentamicin only exhibited ototoxicity. In
addition, the efficiency of dystrophin positive fibers in the mdx mice injected
with hybrid liposome encapsulated gentamicin was higher than in the gentamicin
only injected mice; serum CK levels remained lower in the hybrid liposome group
than in the gentamicin only group. Therefore, the results suggest that the
hybrid liposome encapsulated gentamicin is more effective for DMD patients than
genamicin alone. In order to resolve the patient identification problem, we have
previously developed a system for identifying candidates that qualify for
aminoglycoside therapy. Fibroblasts from 9 DMD patients with nonsense mutations
of the dystrophin gene were isolated, induced to differentiate to myogenic
lineage by AdMyoD, and exposed to gentamicin. The dystrophin expression in
gentamicin exposed myotubes was monitored by in vitro dystrophin staining and
western blotting analysis. The results showed that the gentamicin therapy is far
more effective for DMD patients that have nonsense mutation TGA than for
patients that have nonsense mutation TAA and TAG.
12) Efficient Expression of the 6 kb
Mini-Dystrophin Gene by Trans-Splicing Adeno-Associated Viral (AAV) Vector
Restores the Entire Dystrophin-Associated Glycoprotein Complex and Reduces
Contraction-Induced Damage in the Mdx Mouse Model of Duchenne Muscular
Dystrophy
Yi Lai, Yongping
Yue, Mingju Liu, Arka Ghosh, John F. Engelhardt, Jeffrey S. Chamberlain,
Dongsheng Duan. Department of Molecular Microbiology and Immunology, University
of Missouri, Columbia, MO; Department of Anatomay and Cell Biology, University
of Iowa; Department of Neurology, University of
Washington
Duchenne muscular dystrophy (DMD) is the most
common lethal muscle disease caused by mutations in the dystrophin gene.
Adeno-associated virus (AAV) has been used to express the 3.8 kb C-terminal
deleted micro-dystrophin gene for DMD gene therapy. Despite amelioration of some
disease-associated changes, the microgene is not fully functional.
Trans-splicing AAV vectors have been developed recently to express the 6 kb .jpg) H2-R19 mini-dystrophin gene, the most competent gene besides
the full-length gene. Our recent studies suggest that the RNA processing
represents a critical barrier in trans-splicing AAV vector. In this study, we
first screened a series of endogenous exon/intron/exon junctions by the RNase
protection assay (RPA). The 56/56/57, 60/60/61 and 63/63/64 junctions hold the
highest theoretic splicing values and may represent the most favorable sites to
split the dystrophin gene. We generated synthetic mini-constructs carrying the
splicing signals from each of above junctions. We also inserted the inverted
terminal repeat junction in the middle of the intron. As a control, we included
the 53/53/54 junction that has a low splicing value. The most efficient splicing
was achieved in the 63/63/64 junction where the ratio of the spliced to
unspliced RNA (S/U ratio) reached 7.1
H2-R19 mini-dystrophin gene, the most competent gene besides
the full-length gene. Our recent studies suggest that the RNA processing
represents a critical barrier in trans-splicing AAV vector. In this study, we
first screened a series of endogenous exon/intron/exon junctions by the RNase
protection assay (RPA). The 56/56/57, 60/60/61 and 63/63/64 junctions hold the
highest theoretic splicing values and may represent the most favorable sites to
split the dystrophin gene. We generated synthetic mini-constructs carrying the
splicing signals from each of above junctions. We also inserted the inverted
terminal repeat junction in the middle of the intron. As a control, we included
the 53/53/54 junction that has a low splicing value. The most efficient splicing
was achieved in the 63/63/64 junction where the ratio of the spliced to
unspliced RNA (S/U ratio) reached 7.1 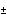 0.9. The
60/60/61 and the 53/53/54 junctions yielded medium S/U ratios of 3.0 0.4
and 2.5 0.3, respectively. The 56/56/57 junction resulted in the lowest S/U
ratio of 1.0 0.6. We also quantified the level of accumulated mRNA.
Surprisingly, the highest mRNA was obtained from the 60/60/61 junction (255.1
20.6, relative unit), followed by the 63/63/64 (139.5 6.4), the
53/53/54 (48.8 20.6) and the 56/56/57 (0.8 0.2)
junctions. We next generated two sets of AAV-6 trans-splicing vectors based on
the 60/60/61 and the 63/63/64 junctions, respectively. 2 x 1010
particles of each set of vectors were delivered to the anterior tibialis (TA)
muscle of 2-m-old mdx mice. Mini-dystrophin expression was quantified at 1 m
post-infection by imunofluorescence staining. Consistent with the RPA results,
trans-splicing vectors based on the 60/60/61 junction yielded 34 fold higher
expression. To further evaluate the therapeutic potential, we delivered the
60/60/61 trans-splicing vectors (donor/acceptor co-infection, or donor or
acceptor single infection) to both the TA and the extensor digitorium longus
(EDL) muscles of the 2-m-old mdx mice. At 3 m post-infection, transduction
efficiency in co-infected TA muscle reached 60-80%. Importantly, the entire
dystrophin-associated glycoprotein complex (including dystroglycan, sarcoglycan,
dystrobrevin and syntrophin) was restored in co-infected muscle. Furthermore,
trans-splicing vector mediated mini-dystrophin expression provided significant
protection against eccentric contraction induced injury in the EDL
muscle.
0.9. The
60/60/61 and the 53/53/54 junctions yielded medium S/U ratios of 3.0 0.4
and 2.5 0.3, respectively. The 56/56/57 junction resulted in the lowest S/U
ratio of 1.0 0.6. We also quantified the level of accumulated mRNA.
Surprisingly, the highest mRNA was obtained from the 60/60/61 junction (255.1
20.6, relative unit), followed by the 63/63/64 (139.5 6.4), the
53/53/54 (48.8 20.6) and the 56/56/57 (0.8 0.2)
junctions. We next generated two sets of AAV-6 trans-splicing vectors based on
the 60/60/61 and the 63/63/64 junctions, respectively. 2 x 1010
particles of each set of vectors were delivered to the anterior tibialis (TA)
muscle of 2-m-old mdx mice. Mini-dystrophin expression was quantified at 1 m
post-infection by imunofluorescence staining. Consistent with the RPA results,
trans-splicing vectors based on the 60/60/61 junction yielded 34 fold higher
expression. To further evaluate the therapeutic potential, we delivered the
60/60/61 trans-splicing vectors (donor/acceptor co-infection, or donor or
acceptor single infection) to both the TA and the extensor digitorium longus
(EDL) muscles of the 2-m-old mdx mice. At 3 m post-infection, transduction
efficiency in co-infected TA muscle reached 60-80%. Importantly, the entire
dystrophin-associated glycoprotein complex (including dystroglycan, sarcoglycan,
dystrobrevin and syntrophin) was restored in co-infected muscle. Furthermore,
trans-splicing vector mediated mini-dystrophin expression provided significant
protection against eccentric contraction induced injury in the EDL
muscle.
13) Recombinant Adeno-Associated Viral (rAAV)
Microdystrophin Vectors as Therapeutic Tools for Duchenne Muscular Dystrophy
(DMD)
Takis Athanasopoulos,
Ian Graham, Helen Foster, Norma Perez, Adelin Vulin, Vanessa Hill, Stewart Fabb,
Luis Garcia, Olivier Danos, George Dickson. 1Centre for Biomedical Sciences,
School of Biological Sciences, Royal Holloway University of London, Egham,
Surrey, United Kingdom; G nthon
III, Evry, Paris, Cedex, France
nthon
III, Evry, Paris, Cedex, France
Duchenne muscular dystrophy
(DMD) is a lethal genetic muscle disorder affecting 1:3500 male individuals,
caused by recessive mutations in the dystrophin gene. The size of the gene
(2.4Mb) and mRNA (14kb) in addition to immunogenicity problems and inefficient
transduction of mature myofibres by currently available vector systems (that
could incorporate the full dystrophin cDNA cassette) are formidable obstacles to
the development of gene therapy. AAV vectors overcome many of the problems
associated with other vector systems but accommodate limited transgene capacity
(<5kb). More than 8 AAV vector serotypes have been identified to date with
certain serotypes (1, 5, 6, 7) displaying more favourable tropism in transducing
muscle fibers compared to the traditionally used AAV2.
A human
microdystrophin cDNA (<3.8 kb) rationally designed in our lab based on
patient and preclinical data incorporated and successfully packaged in an AAV-2
based vector cassette, under the control of a CMV promoter, effectively restored
DAP complex and inhibited myofibre degeneration after local intramuscular
administrations in nude/mdx mice, as reported previously. Other human
microdystrophin versions have been developed in various labs worldwide
displaying different degrees of functionality.
However certain issues
including optimization of transduction, safety and immunogenicity
vector/transgene profiling and species and tissue (muscle) specificity still
remain to be fully addressed. Here we report data on production of human, canine
and murine microdystrophin cDNA constructs via PCR-mediated overlap extension
synthesis, incorporating either constitutive or muscle-specific promoter
elements and optimal Kozak sequences for transcriptional improvement and
packaging of the cassettes into recombinant AAV vectors of alternative serotypes
(1, 5, 6) by pseudotyping. These vectors are currently fully screened and their
functionality determined/compared by extensive in vitro and in
vivo studies utilising local and systemic routes of administration (i.m.,
i.p., i.v. and intra-arterial) in dystrophic and non-dystrophic preclinical
models.
14) Evaluation of Immune Responses to
Dystrophin in Humanized mdx HLA-A*0201 Dystrophic Mice: Application in Duchenne
Muscular Dystrophy Gene Therapy after U7 snRNA-Mediated Exon
Skipping
Florent Ginhoux,
Sabrina Turbant, Marylène Leboeuf, David A. Gross, François Lemonnier, Luis
Garcia, Olivier Danos, Jean Davoust. Immunology, Genethon, Evry, France;
Immunology, Institut Pasteur, Paris, France
Cellular immune
responses may compromise long-term expression in Dystrophin-based gene therapy
treatments. To predict cytotoxic T-cell responses mediated by dystrophin (DYST),
we designed a new H-2-negative HLA-A*0201 transgenic mouse model breaded on mdx
background bearing a stop mutation in DYST exon 23. After DYST plasmid injection
we identified a shared mouse/human specific DYST epitope located on exon 24,
which elicits HLA-A*0201-restricted cytotoxic T cell activities in all mice. We
found however, that rescue of dystrophic muscle by a single administration of an
AAV vector expressing antisense sequences linked to a modified U7 small nuclear
RNA (Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan JC, Garcia L, Danos
O., Science. 2004, 306 : 1796-9) restores long term expression of DYST in muscle
fibres and does not induce CTL activity against DYST in mdx HLA-A*0201 mice.
This persistent exon skipping removes exon 23 and restores DYST exon 24 on the
dystrophin messenger mRNA of the mdx HLA-A*0201 mouse. Several procedures were
then attempted to induce muscle inflammation secondary to exon skipping. They
failed to trigger effector CTL responses and failed to trigger immune rejection
of rescued muscles fibres. We conclude that in contrast to delivery of DYST
plasmid, rescuing dystrophic muscle through U7 snRNA-mediated exon skipping does
not induce a primary immune response directed by exon 24 DYST sequence. The
knowledge of HLA-A*0201-restricted human DYST peptides will be of importance to
assay the occurrence of immune responses in HLA-A*0201 positive humans enrolled
in future clinical studies.
15) Successful Myoblast Transplantation to
Duchenne Muscular Dystrophy Patients
Jacques P. Tremblay, Daniel Skuk, Sylvain Michel,
Raynald Roy, Jean-Guy Lachance, Hélène Senay, Deschênes Louise, Goulet Marlyne,
Brigitte Roy, Jean-Pierre Bouchard. Gntique Humaine, Centre de Recherche du CHUL, Qubec, QC,
Canada; Nphrologie, CHUQ Pavillon Hotel Dieu, Qubec, QC,
Canada; Neurology, Centre Hospitalier Affili, Qubec,
QC, Canada
Nine Duchenne muscular dystrophy (DMD) patients
received injections of myoblasts obtained from skeletal muscle biopsies of
normal donors. Cells were injected in 1 cm3 of the Tibialis anterior
by 25 or 100 parallel injections. We performed similar patterns of saline
injections in the contralateral muscles as controls. The patients received
tacrolimus for immunosuppression. Muscle biopsies were performed at the injected
sites 4 weeks later. We observed dystrophin-positive myofibers in the
cell-grafted sites: in 8 of of 9 patients. In these 8 patients, the percentages
of dystrophin positive fibers ranged from 3.5 to 26%. Eight patients had
identified dystrophin-gene deletions and thus for four of these patients these
results were obtained using mAbs specifically to epitopes coded by the deleted
exons. Donor-dystrophin was absent in the control sites. For the other patients
there were several folds more dystrophin-positive myofibers in the cell-grafted
muscle than in the control muscle. Donor-dystrophin transcripts were detected by
RT-PCR (using one primer reacting with a sequence in the deleted exons) in 8
patients. No antibodies were detected in the host serum against the donor
myoblasts. Therefore, significant dystrophin expression can be obtained in the
skeletal muscles of DMD patients following specific conditions of cell delivery
and immunosuppression.
16) Highly Efficient Exon-Skipping and
Sustained Correction of Muscular Dystrophy Using an Adeno-Associated Viral
Vector
Aurelie Goyenvalle,
Adeline Vulin, Jean Claude Kaplan, France Leturcq, Olivier Danos, Luis Garcia.
DMD Laboratory, Genethon & CNRS UMR8115, Evry, France; Laboratoire de
Biochimie et de Gntique Molculaire, Hopital Cochin, Paris, France
In Duchenne
Muscular Dystrophy (DMD), the lack of Dystrophin results in a series of
catastrophic events that lead to muscle fiber death and muscle wasting.
Dystrophin is a modular protein with a central rod domain composed of 24
spectrin-like repeats and truncated proteins missing some of these repeats can
be fully functional or at least partly deficient as seen in patients with mild
(Becker) forms of DMD. In a majority of severe DMD patients that presents out of
frame alterations of the dystrophin transcript, a translational open reading
frame would be restored by eliminating selected exon(s) from the pre mRNA. Exon
skipping naturally occurs during dystrophin mRNA processing giving rise to rare
revertant fibers that contain shortened proteins. It can be forced using
antisense sequences delivered as synthetic oligonucleotides, or attached to a
small nuclear (sn) RNA, notably U7. A double-target U7SmOPT
containing antisense sequences from introns 22 and 23 (U7-SD23/BP22 ) was
introduced in an AAV vector and packaged as an AAV2/1 pseudotype. Adult mdx mice
were injected into the Tibialis Anterior, or submitted to whole limb arterial
perfusion, and analysed between 2 and 13 weeks. Dystrophin transcripts missing
exon 23 were detected by RT-PCR, representing 15% of the amplified material at
two weeks and becoming the major species (60 to 70%) at one month. The
Dystrophin protein was readily detected both by Western blot on muscle extracts
and by immunofluorescence on tissue sections were restoration of the Dystrophin
Associated Complex was also documented. The levels of rescued dystrophin was 3 %
of normal at 2 weeks and 50 to 80% thereafter. The treated muscles were shown to
recover normal contractile and mechanical properties by measuring resistance to
eccentric contractions. Fiber resistance to exercise-induced damage was also
restored, as seen using Evans Blue exclusion. The level and stability of
U7-induced exon skipping that we observe in vivo in the context of an AAV vector
is unparalleled, and provides a strong rational for the development of this
approach in the clinic.
17) Lentivirus Mediated Dystrophin Expression
in mdx Muscles
En
Kimura, Sheng Li, Brent Fall, Miki Haraguchi, Leonard Meuse, Jeffery S.
Chamberlain. Department of Neurology, University of Washington School of
Medicine, Seattle, WA; Medicine and Biochemistry, University of Washington
School of Medicine, Seattle, WA; Senator Paul D. Wellstone Muscular Dystrophy
Cooperative Research Center, University of Washington School of Medicine,
Seattle, WA
In dystrophic muscles of the mdx mouse,
a model for Duchenne muscular dystrophy, activated-satellite cells (myoblasts)
proliferate in the muscle micro- environment during regeneration and
degeneration processes. Most of these cells terminally differentiate and form
myofibers, while small numbers are stored adjacent to myofibers as mitotically
quiescent satellite cells for future muscle regeneration.
Satellite cells and
other stem cells are an attractive target for genetic modification by
lentiviral vectors, since these vectors enable stable gene expression of a
transgene following integration into the host cells genomic DNA.
We have generated a series of lentiviral vectors that express various reporter
genes and truncated mini- and micro- version of the dystrophin gene, and have
tested their transduction ability in various myogenic precursors both in
vitro and in vivo. Skeletal muscles of newborn mice may be considered
a good target for lentiviral vectors, because of the relatively high numbers of
activated muscle progenitor cells contributing to muscle growth and satellite
cell pools. Therefore, we tried targeting satellite cells or muscle progenitor
cells using lentiviral vectors expressing dystrophin micro-genes using several
different approaches.
Much higher levels of transduction were obtained
following intramuscular injection into neonatal muscles, which are rich with
active myoblasts, compared with muscles from young and adult mice; both muscle
fibers and primary cultured satellite cells stably expressed a GFP marker gene
delivered via lentiviral vectors. Injection of lentiviral vectors
expressing a functional micro-dystrophin/eGFP fusion protein into neonatal
mdx muscles resulted in stable expression of dystrophin for at least one
year, the longest time point current analyzed. Functional studies of the effects
of this transduction are in progress. These data show that myogenic stem cells
can be stably transduced with lentiviral vectors and can contribute to stable
muscle regeneration in dystrophic muscle by enabling continuous expression of
micro-dystrophin, which may have implications for gene therapy of Duchenne
muscular dystrophy.
18) Plasmid-Mediated Gene Transfer in
mdx Mice Using Mini- and Micro-Dystrophin
Constructs
Leland E. Lim,
Carmen Bertoni, Thomas A. Rando. Neurology and Neurological Sciences, Stanford
University School of Medicine, Stanford, CA; Geriatrics Research, Education, and
Clinical Center, VA Palo Alto Health Care System, Palo Alto,
CA
The mdx mouse is a homologue of Duchenne muscular
dystrophy (DMD), which is due to mutations in dystrophin, a subsarcolemmal
protein associated with the actin cytoskeleton. Though the pathological changes
observed in the mdx mouse are different from those of human DMD, the
mdx mouse is an excellent model in which to study potential gene therapy
approaches. One technique is the use of naked
plasmid DNA as a method of gene transfer. Previous work has shown that
dystrophin can be restored to the sarcolemma by intramuscular injection of a
full-length dystrophin cDNA construct. However, expression has been limited to a
small percentage of fibers close to the injection site. Furthermore, the large
size of the dystrophin cDNA presents obstacles to the design of effective
vectors and efficient uptake into muscle fibers. Here, we report the use of two
truncated dystrophin plasmid constructs introduced using electroporation into
the tibialis anterior muscles of young mdx mice. These constructs have
large deletions of the central rod domain while preserving the critical N- and
C-terminal regions. We tested whether these truncated constructs would be more
efficacious than the full-length dystrophin cDNA by virtue of improved delivery
and uptake. In order to follow the expression of dystrophin over time in living
mice, we used a luciferase reporter construct introduced into muscle along with
the dystrophin constructs. Luciferase activity can be followed non-invasively
using a bioluminescent imaging system, and the persistence of luciferase
activity in mdx mouse muscle is dependent upon the expression of
dystrophin to prevent muscle fiber breakdown and concomitant loss of luciferase
plasmids. Our results show that the truncated dystrophin constructs are at least
as effective as full-length dystrophin in preserving luciferase activity. Also,
sarcoglycan and dystroglycan components are present at the sarcolemma of the
dystrophin-expressing fibers, indicating that the truncated dystrophin
constructs are capable of restoring a functional dystrophin-glycoprotein
complex. Current analyses are focusing on the relative distribution of plasmids
in muscle to compare delivery of constructs of different sizes. We are also
analyzing the effectiveness of dystrophin expression by assessing central
nucleation. Finally, our current studies suggest that the longitudinal
distribution along the length of individual fibers is critical to its ability to
protect that fiber over the long term from the degenerative process. At this
stage, we conclude that the smaller dystrophin proteins are effective in
ameliorating the dystrophic features of mdx muscle fibers, and may be
superior to full length dystrophin constructs for the purpose of
plasmid-mediated gene transfer for the treatment of Duchenne muscular
dystrophy.
19) Canine Mini-Dystrophin Gene Transfer by
AAV1 in mdx Mice Ameliorates Dystrophic Pathology and Protects Membrane
Integrity
Bing Wang, Juan
Li, Liqiao Zhou, Chunping Qiao, Mengnan Tian, Tong Zhu, Janet Bogan, Joseph
Kornegay, Xiao Xiao. Dept. of Molecular Genetics and Biochemistry and Dept. of
Orthopedic Surgery, University of Pittsburgh, Pittsburgh, PA; Department of
Veterinary Medicine and Surgery, University of Missouri, Columbia,
MO
Duchenne muscular dystrophy (DMD) is the most common and
lethal genetic muscle disorders, affecting one in 3,500 males. No efficacious
treatment is currently available for DMD. While the mdx mouse is the most
widely used small animal model for DMD, the large animal model, golden retriever
muscular dystrophy (GRMD) dog, displays more similarities to human DMD patients
in clinical and pathological characteristics. Therefore, the GRMD dog is
considered a clinically more relevant and therapeutically more challenging DMD
model.
To investigate AAV-mediated gene therapy for DMD in the canine model,
we have cloned and constructed a canine version of mini-dystrophin gene. The
dystrophin cDNA was partially cloned via RT-PCR from normal dog muscle. The
N-terminus, 5 central rod domains and the cysteine-rich domain were spliced
together, generating the canine mini-dystrophin gene of 3.8 kb, which was
subsequently cloned into the AAV vector driven by the CMV promoter. Before
testing the mini-dystrophin in dogs that have limited availability, we first
tested the construct in the mdx mice to see if the primary biological
functions could be observed.
We show that the canine mini-dystrophin gene is
efficiently expressed in both neonatal and adult mdx mice at one to three
months after AAV1 vector injection. Immunostaining of serial muscle
thin-sections, using antibodies against rods 1 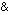 2 region of the
dystrophin and against a, b, and g sarcoglycans separately, revealed the
expression of canine minidystrophin gene and the restoration of the missing
sarcoglycans at the plasma membrane. Importantly, the missing nNOS associated
with myofiber membrane was also partially restored and coincided
mini-dystrophin. H E staining of the vector-treated muscle displayed normal
histology and the lack of fibrosis and infiltration, when compared to the
contralateral untreated gastrocnemius. We found that the central nuclei have
been reduced from 70% to 25% in adult mdx group, and over 98% of nuclei
were peripherally located in areas of young mdx muscle expressing canine
mini-dystrophin. Finally, in vivo muscle cell membrane integrity test after
Evans blue injection also showed improvement and the exclusion of EBD dye
leakage in mini-dystrophin-positive areas. Thus, the results obtained in
mdx mice pave the way for further testing the canine mini-dystrophin in
the GRMD dog model.
2 region of the
dystrophin and against a, b, and g sarcoglycans separately, revealed the
expression of canine minidystrophin gene and the restoration of the missing
sarcoglycans at the plasma membrane. Importantly, the missing nNOS associated
with myofiber membrane was also partially restored and coincided
mini-dystrophin. H E staining of the vector-treated muscle displayed normal
histology and the lack of fibrosis and infiltration, when compared to the
contralateral untreated gastrocnemius. We found that the central nuclei have
been reduced from 70% to 25% in adult mdx group, and over 98% of nuclei
were peripherally located in areas of young mdx muscle expressing canine
mini-dystrophin. Finally, in vivo muscle cell membrane integrity test after
Evans blue injection also showed improvement and the exclusion of EBD dye
leakage in mini-dystrophin-positive areas. Thus, the results obtained in
mdx mice pave the way for further testing the canine mini-dystrophin in
the GRMD dog model.
20) An AAV Vector-Mediated Gene Transfer into
Canine Skeletal Muscle
Madoka Ikemoto, Katsutoshi Yuasa, Madoka Yoshimura,
Akiyo Nishiyama, Yuko Miyagoe-Suzuki, John McC Howell, Shin'ichi Takeda.
Department of Molecular Therapy, National Institute of Neuroscience, NCNP,
Kodaira, Tokyo, Japan; Faculty of Pharmacy, Musashino University, Nishi-tokyo,
Tokyo, Japan; Division of Veterinary and Biomedical Sciences, Murdoch
University, Perth, Western Australia, Australia
Duchenne
muscular dystrophy (DMD) is an X-linked, lethal muscle disorder caused by
mutations in the dystrophin gene (14 kb cDNA). An adeno-associated virus (AAV)
vector-mediated gene transfer is one of attractive approaches to the treatment
of DMD, but it has a limitation in insertion size up to 4.9 kb. We recently
demonstrated that the AAV vector-mediated micro-dystrophin cDNA transfer could
ameliorate dystrophic phenotypes in skeletal muscles of dystrophin-deficient
mdx mice (Mol Ther 10: 821-828, 2004). For clinical application, it is
important to examine therapeutic effects and the safety issue in larger animal
models, such as dystrophic dogs. We established a colony of beagle-based canine
X-linked muscular dystrophy in Japan (Exp Anim. 52: 93-97, 2003). To investigate
transduction efficiency in canine skeletal muscle using an AAV vector, we
injected the AAV vector encoding the LacZ gene driven by a CMV promoter (1.0-2.0
x 1013 vg/ml, 100-500  l/muscle) into
skeletal muscles of normal dogs.
l/muscle) into
skeletal muscles of normal dogs.  -galactosidase
(-gal) was expressed only in few fibers at 2 weeks after the injection,
and not detected at 4 or 8 weeks after the injection. Instead, large numbers of
mononuclear cells appeared around -gal-expressing
fibers. To clarify mechanisms of low transduction and cellular infiltration in
canine muscle after transfer of the AAV vector, we examined viral infectivity
in vitro, cytotoxicity and immune responses of AAV vector transduction
in vivo. First, we infected the AAV vector into canine primary myotubes.
This in vitro study showed that the AAV vector could allow higher
transgene expression in canine myotubes than in murine ones. Second, we tested
whether injection of AAV particles elicit cytotoxicity or not. When a
promoter-less AAV vector expressing no transgene (5 x 1012 vg/muscle)
was injected into canine muscle, almost no infiltrating cells was observed in
injected muscle. Third, we investigated immune responses. A lot of CD4- or
CD8-positive cells were detected in clusters of infiltrating cells, together
with elevated serum level of anti--gal IgG. To
confirm low transduction depending on immune response, dogs received daily oral
administration of cyclosporine (20 mg/kg/day) from
-galactosidase
(-gal) was expressed only in few fibers at 2 weeks after the injection,
and not detected at 4 or 8 weeks after the injection. Instead, large numbers of
mononuclear cells appeared around -gal-expressing
fibers. To clarify mechanisms of low transduction and cellular infiltration in
canine muscle after transfer of the AAV vector, we examined viral infectivity
in vitro, cytotoxicity and immune responses of AAV vector transduction
in vivo. First, we infected the AAV vector into canine primary myotubes.
This in vitro study showed that the AAV vector could allow higher
transgene expression in canine myotubes than in murine ones. Second, we tested
whether injection of AAV particles elicit cytotoxicity or not. When a
promoter-less AAV vector expressing no transgene (5 x 1012 vg/muscle)
was injected into canine muscle, almost no infiltrating cells was observed in
injected muscle. Third, we investigated immune responses. A lot of CD4- or
CD8-positive cells were detected in clusters of infiltrating cells, together
with elevated serum level of anti--gal IgG. To
confirm low transduction depending on immune response, dogs received daily oral
administration of cyclosporine (20 mg/kg/day) from  5 day of the
introduction of the AAV vector. Immunosuppression largely but not completely
improves transduction efficiency of the AAV vector. These results suggest that
AAV vector-mediated gene transfer elicits stronger immune responses in canine
muscle, but immune responses against transgene products can not thoroughly
explain the phenomenon. Cellular toxicity of transgene products might also
participate in these infiltrations, while cytotoxicity and immunity of the AAV
particles themselves can be negligible based on the result of a promoter-less
AAV vectors. It is indispensable to know the molecular background of excess
immune responses and cellular toxicity in canine models to establish AAV
vector-mediated gene transfer in dystrophic patients.
5 day of the
introduction of the AAV vector. Immunosuppression largely but not completely
improves transduction efficiency of the AAV vector. These results suggest that
AAV vector-mediated gene transfer elicits stronger immune responses in canine
muscle, but immune responses against transgene products can not thoroughly
explain the phenomenon. Cellular toxicity of transgene products might also
participate in these infiltrations, while cytotoxicity and immunity of the AAV
particles themselves can be negligible based on the result of a promoter-less
AAV vectors. It is indispensable to know the molecular background of excess
immune responses and cellular toxicity in canine models to establish AAV
vector-mediated gene transfer in dystrophic patients.
21) Overlapping Adeno-Associated Viral (AAV)
Vector Mediated Gene Transfer Is Dependent on Viral Serotype and the Transgene
Sequence in Skeletal Muscle
Arkasubhra Ghosh, Yongping Yue, Dongsheng Duan.
Department of Molecular Microbiology and Immunology, University of Missouri,
Columbia, MO
The small packaging capacity is one of the
major hurdles for adeno-associated virus (AAV)-mediated gene therapy. The
overlapping approach has been developed recently to expand the AAV packaging
capacity (Duan et al, Mol. Ther. 4:383, 2001; Halbert et al, Nat.
Biotechnol.20:697, 2002). In this approach, a large gene is split into two
partially overlapping fragments and separately packaged into two AAV vectors,
including the upstream (carrying the 5-end of the
gene) and the downstream (carrying the 3-end of the
gene) vectors. Transgene expression is achieved after homologous recombination
of the overlapping region between the upstream and the downstream vectors in
co-infected cells. Despite the promising proof-of-principle results in the lung
with AAV-6 alkaline phosphatase (AP) overlapping vectors, the transduction
efficiency in skeletal muscle has been very disappointing. With AAV-2 LacZ
overlapping vectors, the efficiency is only 0.37% of that from an intact AAV-2
LacZ vector. This level of expression is far from sufficient to treat muscular
dystrophy. In this study, we examined two potential rate-limiting factors in the
overlapping approach, including AAV serotype and the transgene sequence.
Previous studies suggest that AAV transduction in muscle is influenced by viral
serotype. AAV-6 mediates much higher gene expression than AAV-2 in muscle. To
test whether AAV-6 can improve overlapping vector-mediated gene transfer in
muscle, we delivered a total of 1 x 10e9 viral genome particles of LacZ
overlapping vectors (AAV-2 or AAV-6; 5 x 10e8 particles of each vector including
the upstream and the downstream vectors) to the anterior tibialis (TA) muscle of
6-week-old BL10 mice. As a control, we also delivered 5 x 10e8 particles of the
intact AAV LacZ vector to the contra-lateral TA muscle. Transduction efficiency
was quantified at 6 weeks later by scoring the percentage of LacZ positive
myofibers. Consistent with previous reports, less than 0.03% of myofibers were
transduced by AAV-2 overlapping vectors. However, AAV-6 overlapping vectors
yielded 35.2 5.7% transduction. This equals 42.25% of that of the
transduction efficiency from an intact AAV-6 LacZ vector. To determine whether
the transgene sequence effects the transduction efficiency, we compared AAV-6
LacZ and AAV-6 AP overlapping vectors. Surprisingly, the transduction efficiency
of AP overlapping vectors (80.4 2.8%) reached
that of the intact AP vector (84.1 1.9%). In
summary, our findings suggest that AAV-6 overlapping vectors represent a
promising approach to deliver certain larger therapeutic genes for muscular
dystrophy gene therapy.
22) Evaluation of Gene Transfer Efficacy
Mediated by AAV1 and AAV6 Vectors in Skeletal Muscle of Adult Mice Following
Different Routes of Administration
Christel Rivière, Karine Poulard, Gabor Veres,
Olivier Danos, Anne M. Douar. Gene Transfert Department - CNRS UMR 8115,
Genethon, Evry, France
Skeletal muscle represents an
important target tissue for gene therapy application due to the large number of
genetic muscular disorders including Duchene Muscular Dystrophy. In addition the
muscle may be also used as a delivery platform to express therapeutic proteins
into the bloodstream for non-muscular metabolic disorders. Adeno-Associated
Virus 1-based vector (rAAV1) is often considered as the most efficient AAV
serotype to deliver genes into the muscles. In this study, we compared the
performance of AAV pseudotyped (based on AAV2 genome) vectors expressing a
secreted form of the murine alkaline phosphatase, (mSEAP) with capsid from
serotype 1 and 6, in mouse skeletal muscle using different delivery methods:
direct injection into individual muscle (intramuscular, IM), local limb
distribution of vectors via the circulation (intra-arterial delivery, IA) or
systemic delivery (intravenous administration, IV). We injected the maximal
volume that each of these route allowed. We analyzed two parameters: the levels
of a reporter circulating protein (), a relevant parameter for a depot organ
strategy, and the extent of fiber transduction in the muscles, a relevant
parameter in view of a muscular disorder therapeutic approach.
We compared
rAAV1 and rAAV6 encoding mSEAP in a dose response study following IM
administration in adult C57Bl/6 mice. Both vectors showed strong and equivalent
levels of transduction at the highest doses (6x108 vg and
3x109 vg), while at the lowest dose injected (1.5x1010
vg), rAAV6 was 3-fold more efficient than rAAV1 in transducing muscle,
suggesting a differential threshold of efficiency at sub-optimal doses.
In
addition, the potential for systemic gene transfer after IV of rAAV1 and rAAV6
at the whole body level was investigated. For the two doses tested
(1x1011 vg and 3x1011 vg). AAV6 vector led to 3-fold
higher levels of circulating mSEAP levels than rAAV1. In addition, a widespread
transduction of both skeletal and cardiac tissues was observed with rAAV6 after
histochemical detection of mSEAP. However, in term of circulating protein level,
IV administration was less efficient than IM injection since the same level of
circulating mSEAP was achieved with 3x1011 vg using IV delivery
compared to3x109 vg for IM delivery.
We have also evaluated
intra-arterial delivery via the femoral artery of rAAV1 and rAAV6 at two doses
(1x1011 vg and 3x1011 vg). This route yielded a 10 fold
higher mSEAP levels in the serum than IV and led to robust transgene expression
pattern in the hind limb muscles.
Finally, in an attempt to increase gene
transfer efficacy in muscle after IV delivery, we co-injected the VEGF, with
either rAAV1 or rAAV6. For both vectors, the presence of VEGF did not show a
positive impact on transgene expression levels.
In conclusion, our results
show that despite the high similarity between AAV1 and AAV6 capsid sequences,
rAAV6 performance for muscle transduction are superior to rAAV1 with all IM, IA
or IV delivery routes. This finding prompts us to consider AAV6 vectors as one
of the most efficient viral agent to deliver gene in skeletal muscle and further
clinical applications.
23) Identification of Differentially
Expressed Genes in Duchenne Muscular Dystropy Utilising RNAi Technology:
Possible Targets for Gene Therapy
Mohammad Mahdi Ghahramani Seno, Ian R. Graham, Ken
Laing, Marita Pohlschmidt, Takis Athanasopoulos, Mark Crompton, George J.
Dickson. Centre for Biomedical Research, Royal Holloway-University of London,
Egham, Surrey, United Kingdom; Bacterial Microarray Group London, St Georges
Hospital Medical School University of
London, London, United Kingdom
Duchenne Muscular Dystrophy
(DMD) is one of a group of genetically heterogeneous muscular dystrophies that
are characterized by progressive weakness and wasting of skeletal muscle. Loss
of myofibres occurs in response to a deficiency of dystrophin, a protein which
is believed to be responsible for myofibre maintenance and integrity. Dystrophin
forms a link between the cytoskeleton and the membrane-spanning
dystrophin-associated glycoprotein complex (DAPC), indicative of a structural
role for dystrophin.
The application of gene therapy protocols for DMD still
presents many daunting challenges due partly to intrinsic features of the
dystrophin gene. Hence, improvement in the understanding of the underlying
primary molecular events leading to a dystrophic pathology might pave the way
for the discovery of new starting points.
Here we present a strategy to use
RNAi technology to study the events occurring in muscle cell development due to
dystrophin deficiency. RNAi has been proven to be a powerful technology to study
molecular effects due to knockdown of single genes. We have used a series of
siRNAs to target and knock down the expression of dystrophin in primary cultures
of mouse muscle, and subsequently used transcriptomic array analysis to identify
genes whose expression was affected in response to dystrophin deficiency. The
data obtained from this experiment, which include some very interesting
potential new targets, are currently being analysed. We are also developing a
recombinant adeno-associated virus (rAAV) vector expressing an shRNA targeting
dystrophin. The use of such rAAV-shDNA vectors will enable us to target
dystrophin in vivo to obtain a better and potentially curative insight into the
pathophysiology of DMD.
24) Perivascular
CD45-:Sca-1+:CD34- Cells Are Derived from Bone
Marrow Cells and Participate in Dystrophic Skeletal Muscle
Regeneration
Sheng Li,
Morayma Reyes, En Kimura, Jessica Foraker, Miki Hagakura, Leonard Meuse, Brent
Fall, Jeffrey S. Chamberlain. Department of Neurology, University of Washington
School of Medicine, Seattle, WA; Muscular Dystrophy Cooperative Research Center,
University of Washington School of Medicine, Seattle,
WA
Multiple mechanisms may account for bone marrow (BM)
cell incorporation into myofibers following muscle damage. Here, we demonstrated
that mouse CD45-:Sca-1+:CD34- cells may play a
role in the maturation of skeletal muscles and regeneration of
mdx4cv dystrophic skeletal muscles, an animal model for
Duchenne muscular dystrophy. To understand the origin of
CD45-:Sca-1+:CD34- cells in mouse skeletal
muscle, we reconstituted lethally irradiated wild type or
mdx4cv mice with unfractionated BM cells from transgenic mice
ubiquitously expressing green fluorescence protein (GFP). 1, 2, and 6 months
post-transplantation, we analyzed the skeletal muscle mononuclear cells from the
recipients by flow cytometry for GFP, CD45-PerCP, Sca-1-PE, and CD34-APC. To our
surprise, we found BM-derived (GFP+)
CD45-:Sca-1+:CD34- cells in the skeletal
muscles of these GFP+ BM transplant recipients. We also demonstrated
that these BM-derived cells were localized in the perivascular tissue by
immuno-staining and that their frequency increased with time. We were interested
in the potential clinical application of these cells for muscle diseases. Thus,
we sorted CD45-:Sca-1+:CD34- cells by
fluorescence activated cell sorting (FACS) skeletal muscle mononuclear cells and
cultured them in several stem cell media (recipes), including a low-serum medium
containing specific cytokines for isolating multipotent adult progenitor cells
(MAPCs). MAPCs can be isolated from skeletal muscle and BM and differentiate to
form myotubes in vitro and in vivo. Strikingly, we found that
MAPCs were enriched up to 40 folds by sorting this population from skeletal
muscle mononuclear cells. Concomitantly with the increase in frequency of
BM-derived muscle CD45-:Sca-1+:CD34- cells,
frequency of BM-derived muscle MAPCs also increased with time
post-transplantation in dystrophic muscles. Furthermore, BM-derived muscle MAPCs
displayed similar phenotypes of endogenous muscle MAPCs, suggesting a potential
mechanism of BM cell migration to dystrophic skeletal muscles. To understand how
these BM-derived cells migrate to the muscle and once in the muscle how they
mobilize, we investigated the in vitro chemotatic response of
GFP+ (BM derived) muscle MAPCs and CD45-:Sca-1+
cells isolated from muscles of GFP+ BM transplant recipients. We
found that these cells were highly chemoattrated to stroma derived factor,
SDF-1, a chemo-attractant for cells expressing CXCR4. We also observed higher
frequency of BM-derived CD45-:Sca-1+:CD34-
cells in mdx dystrophic muscle than wild type muscle, which may be
explained by higher expression levels of SDF-1 in mdx dystrophic muscles.
Taken together, our results suggest that dystrophic muscles recruit BM cells
that localize in perivascular tissues and can be defined as
CD45-:Sca-1+:CD34-. This population when
cultured enriches for MAPCs and can participate in muscle regeneration in
dystrophic muscles.
25) AAV-Mediated Myostatin Propeptide Gene
Delivery Results in Growth and Hypertrophy of Skeletal but Not Cardiac
Muscles
Chunping Qiao,
Jianbin Li, Tong Zhu, Chunlian Chen, Terry O'Day, Jon Watchko, Juan Li, Xiao
Xiao. Department of MGB, University of Pittsburgh, Pittsburgh, PA; Dept. of
Pediatrics, University of Pittsburgh, Pittsburgh, PA; Dept. of Orthopaedic
Surgery, University of Pittsburgh, Pittsburgh, PA
Muscular
dystrophies are inherited myogenic disorders characterized by progressive muscle
wasting and weakness. Because of the lack of effective treatment with
traditional pharmaceutical agents, gene therapy and stem-cell therapy have been
vigorously explored. Besides gene replacement therapy, a different form of gene
therapy, i.e., expression of booster
genes, has been investigated, aiming at alleviating the secondary
deficiencies in muscular dystrophies rather than the primary ones. Blockade of
myostatin is one of such strategies. Myostatin is a member of the TGF-beta
family and a negative regulator of skeletal muscle growth. Studies using
transgenic mice or antagonist antibody against myostatin showed promotion of
muscle growth and amelioration of muscular dystrophy. However, gene delivery of
myostatin inhibitors has not been reported.
In this study we examined
whether myostatin propeptide gene transfer into normal mice by AAV vectors could
increase muscle mass and strength. AAV vector carrying myostatin propeptide
(MPRO) gene was delivered into neonate and adult BL10 mice respectively. For
neonates study, AAV serotype 8 carrying MPRO gene was delivered into 3 to 5 days
old mice by intraperitoneal injection. Two months after vector injection we
observed very significant hypertrophy of the skeletal muscles. The treated mice
gained body weight and had less fat when compared to the controls. The skeletal
muscle mass including TA, GAS, Quadriceps, and Diaphragm from the treated mice
were significantly larger (p<0.01). Myofiber diameters of the treated mice
were larger than the untreated ones. The hypertrophy was not observed in the
hearts of the treated mice despite the gene expression there. However, the
larger muscle did not yield stronger force. The peak twitch and peak tetanic
force of the treated muscle did not increase significantly, and the treated mice
also did not increase performance on the treadmill. For the adult study,
AAV-MPRO gene was delivered by local and systemically. Local intramuscular
injection study indicated that the treated muscle mass also increased
significantly. However, the treated muscle showed increased central nucleation,
suggesting muscle regeneration. The study of systemically delivery of AAV-MPRO
gene to the adult mice is under way. In conclusion, these data have shown for
the first time that muscle mass can be increased by gene delivery of myostatin
propeptide in normal mice. Further studies on muscular dystrophy animal models
are warranted.
26) Real Time Imaging of Myoblast
Transplantation Using the Human Sodium Iodide Symporter (hNIS) as Reporter
Gene
Manaf Bouchentouf,
Basma F. Benabdallah, Joel Rousseau, Marcel Dumont, Jacques P. Tremblay. Human
Genetic unit, CHUL, Ste-Foy, QC, Canada; Radiology Unit, HSFA, Quebec, QC,
Canada
BACKGROUND: The quantification of the graft
success is a key element to evaluate the efficiency of cellular therapies for
several pathologies such as Duchenne muscular dystrophy. This study describes a
novel approach to evaluate the success of myoblast transplantation (i.e., the
survival or the transplanted cells and of the muscle fibers that they have
formed) by real-time imaging. METHODS: C2C12
myoblasts were first transfected with a plasmid containing the human sodium
iodide symporter (hNIS) gene. The specific uptake of
99mTCO4 by the hNIS positive myoblasts was demonstrated
in vitro, no accumulation of 99mTCO4 was observed
within the control cells. The cells were then transplanted into the Tibialis
anterior (TA) muscle of mdx mice. Following intra-peritoneal
administration of Na99mTCO4, scintigraphies
(g-radiographs) were performed depicting hNIS-dependent
99mTCO4 uptake within the TA. RESULTS:
Preliminary experiments have demonstrated that the resolution of the camera used
is 2 mm. In vitro the cells expressing the hNIS were able to incorporate
the Na99mTCO4 and were also able to
form myotubes. In vivo image acquisition of transplanted mice
TA revealed that hNIS positive cells were able to incorporate the
Na99mTCO4. CONCLUSIONS: This approach permitted to
evaluate the progression of the transplantation and the graft success without
having to biopsy the animals during the follow-up period.
27) Overexpression of the Myostatin
Antagonist Follistatin in Normal Myoblasts Genetically Modified with a
Lentivirus
Basma F.
Benabdallah, Joel Rousseau, Manaf Bouchentouf, Jacques P. Tremblay. Human
Genetic, CHUL-CHUQ-Laval University, Sainte Foy, QC,
Canada
Background: Duchenne muscular dystrophy is a
severe myopathy caused by the absence of a functional dystrophin in muscles.
Transplantation of normal myoblasts is a potential therapy that permits to
restore the expression of the dystrophin in transplanted muscles by the
formation of hybrid dystrophin positive fibers. However, the success of this
approach is compromised by the limited regeneration of damaged muscle.
Myostatin, the most powerful inhibitor of muscle growth identified to date, is
regulated by different antagonist proteins such as follistatin. Our purpose is
to block the myostatin signal in mdx and SCID host mice by the respective
transplantation of normal murine and human myoblasts genetically modified with a
follistatin lentivirus.
Methods: 293T packaging cell line was used to
produce a lentivirus coding for the short form of follistatin protein under the
control of a cytomegalovirus promoter. Normal murine and human myoblasts were
infected with the lentivirus to overproduce the follistatin protein. The
surexpression of the follistatin in both transfected 293T cells and infected
murine and human normal myoblasts was verified by immuno-cytochemistry assay and
Western blot. A lentivirus containing the eGFP gene was used as a control. The
differentiation of control and lentivirus-follistatin infected myoblasts was
also evaluated by the determination of the fusion index.
Results: The
immunocytochemistry assay showed that the follistatin was clearly overexpressed
in both pCMV-Fst transfected 293T cells and infected murine and human normal
myoblasts compared with the control cells transfected or infected with the eGFP
lentivirus. The Western blot also demonstrated that the follistatin was
effectively overexpressed in pCMV-Fst transfected 293T cells and infected murine
and human normal myoblasts compared with the control cells transfected or
infected with the eGFP lentivirus. The fusion index test also showed that
myoblasts infected with the follistatin lentivirus formed more myotubes than
myoblasts infected with the eGFP-lentivirus when cultured in differentiation
conditions.
Conclusions: The transplantation of normal murine or human
myoblasts overexpressing follistatin, in mdx or SCID mouse respectively
in order to antagonize the myostatin signaling, could be a good approach to
improve the success of the potential cellular therapy of Duchenne
myopathy.
28) Development of AAV-Mediated Gene Therapy
for Murine Models of Genetic Diseases Affecting the
Heart
Christina A. Pacak,
Cathryn Mah, Gabriel Gaidosh, Melissa Lewis, Raquel Torres, Kevin Campbell,
Glenn A. Walter, Barry J. Byrne. Molecular Genetics and Microbiology, University
of Florida, Gainesville, FL; Powell Gene Therapy Center, University of Florida,
Gainesville, FL; Physiology, University of Florida, Gainesville, FL; Cellular
and Molecular Therapy, University of Florida, Gainesville, FL; HHMI, University
of Iowa, Iowa City, IA
The long term goal of this project
is to develop a clinically relevant gene therapy approach for the treatment of
genetic diseases affecting the heart. Due to its small size, safety and proven
ability to persist for long periods of time in muscle, adeno-associated virus
(AAV) has emerged as a promising cardiac gene delivery vehicle. We have sought
to determine the most advantageous combination AAV serotype and vector delivery
route for the transduction of cardiomyocytes in vivo. Both intra-venous
(iv) and intra-cardiac (ic) injection routes were compared by injecting
1x1011 and 5x1010 (respectively) vector particles of
AAV-CMV-LacZ per mouse neonate of 3 different serotypes AAV1, AAV8 and AAV9.
Tissue analysis included both x-gal staining on tissue sections to visualize
expression and the quantitative -galactosidase
enzyme detection assay. Our results show that iv administration of AAV9 results
in 30-fold more efficient transduction of cardiac tissue than AAV1. Moreover,
hearts injected with AAV9 displayed a global distribution of transgene
expression suggesting this serotype has no transduction site preference within
cardiac tissue. 2 murine models for cardiac dysfunction; the alpha-sarcoglycan
(asg -/-) knockout model for Limb Girdle Muscular Dystrophy
Type 2D (LGMD-2D) and the acid-alpha glucosidase (gaa-/-)
knockout model for Pompe Disease are currently being characterized functionally
and morphologically using both non-invasive MRI and histological techniques.
LGMD-2D is the result of mutations in the alpha sarcoglycan (ASG) gene. This
model displays the development of dystrophic lesions in cardiac and skeletal
muscle. We have developed a non-invasive assay using MRI that enables us to
locate and measure the random development of lesions within the muscles of
asg -/- mice. We have demonstrated functional correction and
prevention of disease progression in skeletal muscles injected with AAV-ASG.
Studies are currently underway to demonstrate the same effect in cardiac tissue.
The cells of the gaa-/- mouse contain lysosomes enlarged with
glycogen due to a GAA enzyme deficiency. Previously, our lab has demonstrated
that delivery of the human GAA gene to the skeletal muscle of this model is
therapeutically beneficial. To evaluate gene delivery to the cardiac tissue of
this model we are currently characterizing the cardiac phenotype of disease and
assessing our ability to prevent disease presentation following optimized
delivery of the human GAA gene. In conclusion, our maker-gene study has
established that iv delivery using AAV9 is the most clinically advantageous
combination of AAV serotype and delivery route for the transduction of cardiac
tissue in vivo. We are now in the process of using this delivery method
to demonstrate disease correction in two mouse models of
cardiomyopathy.
29) Decorin Promotes Differentiation of
Myoblasts into Myotubes That Express Slow MyHC In Vitro
Yong Li, Juan Li, Ying Tang, Xiao Xiao, Johnny
Huard. Dept. of Orthopaedic Surgery, Childrens Hospital and
University of Pittsburgh, Pittsburgh, PA; Dept. of Molecular Genetic and
Biochemistry, University of Pittsburgh, Pittsburgh,
PA
Decorin, a small leucine-rich proteoglycan, is a key
regulator in extracellular matrix assembly and cell proliferation. It also has a
specific effect on the migration and differentiation of embryonic skeletal
muscle cells (1). We have demonstrated that decorin can prevent muscle fibrosis,
enhance muscle regeneration, and improve the functional recovery of injured
skeletal muscle (2). However, the mechanism behind decorins effect on
muscle regeneration is unclear. In the current study, we used an AAV-mDecorin
plasmid to transfect a mouse decorin gene into myoblasts (C2C12 cells) and
evaluated the phenotypic changes during the resultant myoblast differentiation.
Our results revealed that myoblasts engineered to express decorin undergo
differentiation more readily than do normal myoblasts in vitro. This enhanced
differentiation may be due to the stimulation of P21, an important
cyclin-dependent kinase inhibitor that is up-regulated during muscle cell
differentiation (3). We also observed up-regulated expression of myogenic
proteins (MyoD, Myogenin, Myf5, and Myf6) by these decorin-transfected (CD)
cells. This study demonstrates that decorin treatment resulted in the
overexpression of slow myosin heavy chain (MyHC) and the production of slow
myotubes by CD cells in vitro. These results suggest that decorin may promote
muscle regeneration through the activity of slow MyHCexpressing
muscle fibers. These results indicate that decorin improves skeletal muscle
healing not only by preventing fibrosis, but also by promoting muscle
regeneration. These findings could explain why decorin treatment can lead to
nearly complete functional recovery of injured muscle (2).
References:
1.
Villena J, et al. J Cell Physiol 2004;198:16978.
2.
Fukushima K, et al. Am J Sports Med 2001;29:394402.
3.
Halevy O, et al. Science. 1995;267:1018-21.